Case Report
A case report of Hypocomplementemic urticarial vasculitic syndrome presenting with Renal failure

Amy M Hopkins1, Angela M Gibbs1, Ryan S Griffiths2, Rupali S Avasare3 and Firas G Khoury2*
1Providence St. Vincent Medical Center, Department of Internal Medicine Portland, Oregon, USA
2Oregon Kidney & Hypertension Clinic, Portland, Oregon, USA
3Oregon Health & Science University, Portland, Oregon, USA
*Address for Correspondence: Dr. Firas G Khoury, MD, The Oregon Kidney & Hypertension Clinic, Portland, Oregon, USA, Email: firas.khoury@davita.com
Dates: Submitted: 03 September 2018; Approved: 11 September 2018; Published: 12 September 2018
How to cite this article: Hopkins AM, Gibbs AM, Griffiths RS, Avasare RS, Khoury FG. A case report of Hypocomplementemic urticarial vasculitic syndrome presenting with Renal failure. J Clini Nephrol. 2018; 2: 039-043. DOI: 10.29328/journal.jcn.1001017
Copyright License: © 2018 Khoury FG, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Hypocomplementemic urticarial vasculitic syndrome; Chronic urticarial; Leukocytoclastic vasculitis; Cryoglobulinemia; Acute renal failure

Abstract
We present a case of hypocomplementemic urticarial vasculitic syndrome (HUVS) who developed severe renal failure requiring ICU-level care. Our patient is a 66-year-old man who presented with abdominal pain, rash, confusion, oliguria, and shortness of breath. He was found to be in acute renal failure with leukocytosis and elevated lactate. Work-up for infectious, autoimmune, and hematologic malignant diseases was negative. The presence of chronic urticaria, abdominal pain, hypocomplementemia, and leukocytoclastic vasculitis on skin biopsy confirmed the diagnosis of HUVS. He required hemodialysis for renal failure as well as gastrostomy tube placement for nutritional support secondary to the development of mucosal ulcers, a rare finding in HUVS. He recovered with several months of high-dose steroids and hemodialysis. This case highlights the effectiveness of steroids for initial treatment of HUVS, and the relapsing and remitting nature of the disease. Providers should also be aware of the broad range of presenting symptoms such as mucosal lesions that may require nutritional support. Interestingly, unlike many previously reported cases of HUVS, our patient had not yet developed signs and symptoms of systemic lupus erythematosus, which often overlaps with HUVS.
Introduction
Hypocomplementemic urticarial vasculitic syndrome (HUVS) is a rare disorder characterized by chronic urticarial vasculitis and hypocomplementemia [1]. First described in 1973 by McDuffie et al. [2], the etiology of HUVS remains controversial [3-8]. HUVS is triggered by an unknown event that leads to immune complex deposition in small blood vessels and activation of the complement pathway. Diagnostic criteria defined by Schwartz et al. in 1982 require presence of both major criteria plus at least two minor criteria for diagnosis (Table 1) [9].
| Table 1: Diagnostic criteria for hypocomplementemic urticarial vasculitic syndrome according to Schwartz et al (1982). |
| Major Criteria (must have both) |
| Urticarial skin lesions |
| Low levels of serum complement |
| Minor Criteria (must have 2) |
| Dermal venulitis |
| Arthritis |
| Glomerulonephritis |
| Episcleritisor uveitis |
| Recurrent abdominal pain |
| C1q precipitin in plasma |
Multi-organ involvement is common including the skin (100% of cases), joints (70%), kidneys (50%), gastrointestinal system (30%), lungs (20%), eyes (10%), and rare involvement of the heart and nervous system [10]. Urticarial vasculitis is a distinct entity that differs from urticaria by erythematous wheals that are less pruritic and more painful, persist for greater than 24 hours, and leave hyperpigmentation after healing [10]. Joint involvement is often in the form of Jaccoud’s arthropathy, migrating polyarthralgia that was classically described in rheumatic fever but has also been described in systemic lupus erythematosus (SLE) and other diffuse connective tissue diseases [10,11]. Renal involvement can range from mild proteinuria or microscopic hematuria to nephrotic syndrome, membranoproliferative glomerulonephritis, or rapidly progressive glomerulonephritis [12-16].
Laboratory measurements of complement levels are necessary for diagnosis (Table 1). Although C3 levels can be variably decreased, C4 levels are often low [4]. Additionally, the majority of patients have auto-antibodies against C1q and low C1q [1,3,4]. The presence of antibodies against C1q supports a type III hypersensitivity reaction with immune complex-mediated injury [17]. Indeed, immunofluorescence of biopsies of involved organs show immunoglobulin and complement deposits in the vessel walls or on the endothelium, which is in contrast to deposition along the glomerular basement membrane seen in SLE [10]. Dermal biopsies of the active rash of HUVS show leukocytoclastic vasculitis with a perivascular neutrophil infiltrate. Antinuclear antibodies and elevated ESR are present 60-70% of the time [4]. Although rheumatoid factor, anti-dsDNA antibodies, and anti-SS-A / SS-B antibodies may be abnormal, they are elevated in only 8-17% of patients with HUVS [4].
Treatment of HUVS depends on the severity of disease. Anti-histamines and glucocorticoids are the cornerstone of treatment to combat inflammation and immune complex formation. Methotrexate, azathioprine, cyclophosphamide, and cyclosporine have also been reported with several cases reporting notable success with mycophenolate mofetil [13-15,10,18,19]. Plasmapharesis will reduce circulating immune complexes, but symptoms return as anti-C1q antibody production continues [10,12]. Dapsone has been reported in pediatric cases, but its role has not been clearly delineated in adults [13].
Case Report
Our patient is a 66-year-old Caucasian male Jehovah’s Witness with a history of diabetes, hypertension, stroke, gout, and hyperlipidemia who presented with confusion, right upper quadrant pain, worsening shortness of breath, and decreased urination for one day. Four days prior, he presented to the emergency room for a rash on his abdomen and lower extremities (Figure 1A,B). Lab results at that time were not concerning for an acute process. He was diagnosed with chronic urticaria and sent home with prednisone. At his next presentation, he was anuric. Labs were significant for a white count of 20 x 109/L, lactate of 5.7mmol/L, BUN of 91mg/dL, and creatinine of 9.14mg/dL, which had increased from 1.09mg/dL four days prior. His urinalysis showed proteinuria and hematuria raising suspicion for glomerulonephritis as the cause of his renal failure. Additional testing showed elevated ESR, CRP, and low CH50. Complement levels were initially low with C3 of 65mg/dL and C4 that was <8mg/dL, normalized while on high dose steroids, and subsequently decreased again upon reduction of steroid dose. Although an early sample sent for cryoglobulin immunofixation electrophoresis was positive for type III cryoglobulinemia, repeat qualitative cryoglobulin testing 3.5 weeks later failed to reveal cryoglobulin formation.
Renal biopsy was initially delayed due to the patient’s critical condition and risks associated with the patient’s refusal of blood products as a Jehovah’s Witness. Five days into hospitalization, a skin biopsy showed leukocytoclastic vasculitis despite urticarial appearance. After clinical stabilization, a kidney biopsy was performed. For light microscopy, three intact glomeruli and portions of a fourth and fifth were examined. There was mild patchy interstitial fibrosis with minimal chronic inflammation in areas of fibrosis. Four to five glomeruli were present in most levels for immunofluorescence evaluation. Staining for IgA, IgG, IgM, fibrinogen, kappa light chains, and lambda light chains was negative. C3 staining yielded 1-2+ mesangial staining. There was insufficient sample size for electron microscopy. The final impression was an unsampled glomerulonephritis given persistent hematuria and proteinuria versus acute tubular necrosis from capillary leak syndrome.
Ultimately, our patient required hemodialysis for his acute renal failure. He also required percutaneous gastrostomy tube placement for nutritional support due to decreased intake secondary to severe mouth sores on the tongue, buccal mucosa, and esophagus thought to be secondary to underlying HUVS. Despite significant deconditioning, he clinically improved on steroids alone via gastrostomy tube. Steroids were tapered from 60mg per day by 10mg every 3 weeks. Dialysis was discontinued after four months of therapy.
Unfortunately, he suffered a relapse of his HUVS two months after his recovery from dialysis and completion of steroid taper. The repeated presentation was evidenced by return of arthralgias, urticaria, confusion, decreased appetite, and acute on chronic renal failure. The patient’s ESR and CRP were also elevated. Prompt oral steroid treatment was started without need for dialysis. His symptoms resolved and his renal function has returned to his prior baseline . Mycophenolate mofetil is being considered for any future relapses.
Discussion
The diagnosis of HUVS in our patient is supported by fulfillment of the two major criteria (chronic urticaria and hypocomplementemia) and two minor criteria (leukocytoclastic vasculitis on skin biopsy and abdominal pain). Autoimmune cryoglobulinemic vasculitis remained on the differential diagnosis for the majority of his hospitalization until repeat testing returned negative after receiving steroids, though we note the limitations of serum cryocrit testing. Other findings that supported HUVS over cryoglobulinemic vasculitis included the presence of chronic urticaria (Figure 1A,B) that resolved with hyperpigmented macules and patches as opposed to palpable purpura expected in cryoglobulinemic vasculitis. His hematologic malignancy work-up also returned negative. Our patient also did not fulfill diagnostic criteria for SLE. According to a retrospective study by Davis et al., 24 out of 132 patients with urticarial vasculitis had hypocomplementemia, 54% of whom developed SLE [20]. Many case reports since have presented patients with both diagnoses of SLE and HUVS [5,7,20,21]. Although understanding of the genetics and pathophysiology of HUVS and SLE is far from complete, providers should be aware of the dual diagnoses to provide appropriate treatment for both conditions.
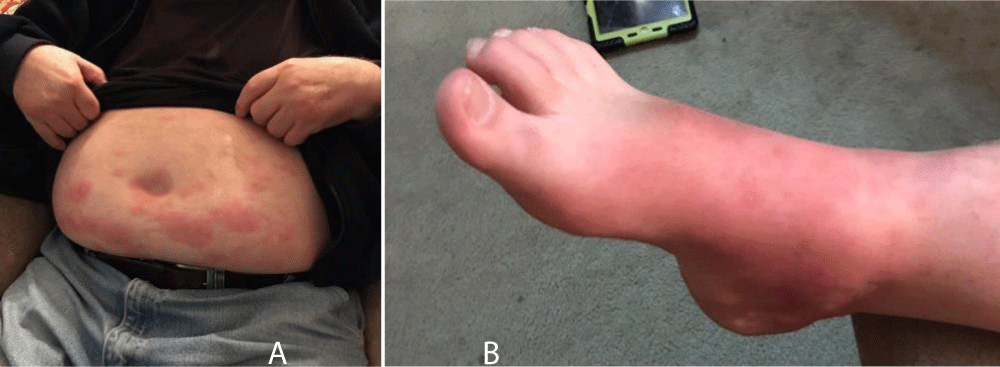
Figure 1: A: Erythematous papules and plaques scattered diffusely across the abdomen of our patient with hypocomplementemic urticarial vasculitis. B: Diffuse erythema on the dorsal aspect of the right foot of the same patient with violaceous and hyperpigmented patches scattered throughout.
Perhaps the most striking clinical feature of our patient’s case of HUVS is his severe renal involvement, most likely glomerulonephritis based on active urine sediment, though this was not confirmed on kidney biopsy due to a very limited biopsy sample. Progression to end-stage renal failure requiring dialysis in HUVS is rare (~10% of cases as reported by Toprak et al.) [12-15]. Diagnosis of glomerulonephritis due to HUVS in our patient is also supported by his improvement with steroids alone, the cornerstone treatment of HUVS . Furthermore, our patient developed oral ulcers. Although the ulcerations were severe enough to require gastrostomy tube placement for nutrition, the lesions resolved with high-dose steroids suggesting that they were also a manifestation of his HUVS. To our knowledge, there are no prior reports of mucosal involvement in HUVS.
Our case highlights the range and severity of organ system involvement possible in HUVS. The variability of presenting symptoms adds to the diagnostic complexity that is already well described in relation to overlap with other systemic diseases including cryoglobulenemic vasculitis, hematologic malignancies, and SLE. High prevalence of the co-existence of HUVS and SLE support close follow-up of patients diagnosed with HUVS for development of signs and symptoms of SLE. As a systemic disease involving multiple organ systems, providers should be aware of serious complications of HUVS such as renal failure and mucosal ulceration. Despite a chronic remitting and relapsing course, management with steroids alone can offer periods of remission with other immunosuppressants such as cyclophosphamide, rituximab, and mycophenolate mofetil as second-line agents in the case of relapse.
Acknowledgement
The authors would like to thank Dr. Shweta S. Patel, MD at The Oregon Kidney & Hypertension Clinic for following up with the patient and providing clinical data that contributed to the manuscript.
References
- Park C, Choi SW, Kim M, Park J, Lee JS, et al. Membranoproliferative glomerulonephritis presenting as arthropathy and cardiac valvulopathy in hypocomplementemic urticarial vasculitis: a case report. J Med Case Rep. 2014;8: 352. Ref.: https://goo.gl/kCxW3U
- McDuffie FC, Sams Jr. WM, Maldonado JE, Andreini PH, Conn DL, et al. Hypocomplementemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin Proc. 1973; 48: 340–348. Ref.: https://goo.gl/aYaFoc
- Aydogan K, Karadogan SK, Adim SB, Tunali S. Hypocomplementemic urticarial vasculitis: a rare presentation of systemic lupus erythematosus. Int J Dermatol. 2006; 45: 1057–1061. Ref.: https://goo.gl/Po97id
- Buck A, Christensen J, McCarty M. Hypocomplementemic urticarial vasculitis syndrome: a case report and literature review. J Clin Aesthet Dermatol. 2012; 5: 36-46. Ref.: https://goo.gl/2uc5R6
- Carbonella A, Mancano G, Gremese E, et al. An autosomal recessive DNASE1L3-related autoimmune disease with unusual clinical presentation mimicking systemic lupus erythematosus. Lupus. 2017; 26: 768–772. Ref.: https://goo.gl/jWmtjm
- Davis MD, Brewer JD. Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am. 2004; 24: 183–213. Ref.: https://goo.gl/DUh8Hn
- Her MY, Song JY, Kim DY. Hypocomplementemic urticarial vasculitis in systemic lupus erythematosus. J Korean Med Sci. 2009; 24:184–186. Ref.: https://goo.gl/1kSQMo
- Wisnieski JJ1, Baer AN, Christensen J, Cupps TR, Flagg DN, et al. Hypocomplementemic urticarial vasculitis syndrome. Clinical and serologic findings in 18 patients. Med. 1995; 74: 24–41. Ref.: https://goo.gl/a7JQrD
- Schwartz HR, McDuffie FC, Black LF, Schroeter AL, Conn DL. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clin Proc. 1982; 57: 231–238. Ref.: https://goo.gl/TpMKSD
- Grotz W, Baba HA, Becker JU, Baumgartel MW. Hypocomplementemic urticarial vasculitis syndrome: an interdisciplinary challenge. Dtsch Arztebl Int. 2009; 106: 756–763. Ref.: https://goo.gl/ckoimH
- Santiago M, Machicado V. IMAGES IN CLINICAL MEDICINE. Jaccoud’s Arthropathy. N Engl J Med. 2015; 373: e1. Ref.: https://goo.gl/1udvsJ
- Grimbert P, Schulte K, Buisson C, Desvaux D, Baron C, et al. Renal transplantation in a patient with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis. 2001; 37: 144–148. Ref.: https://goo.gl/rcZFMN
- Pasini A, Bracaglia C, Aceti A, Vivarelli M, Lavacchini A, et al. Renal involvement in hypocomplementaemic urticarial vasculitis syndrome: a report of three paediatric cases. Rheumatol. 2014; 53: 1409–1413. Ref.: https://goo.gl/CezVFX
- Salim SA, Yousuf T, Patel A, Fulop T, Agarwal M. Hypocomplementemic Urticarial Vasculitis Syndrome With Crescentic Glomerulonephritis. Am J Med Sci. 2018; 355: 195–200. Ref.: https://goo.gl/yK6tD6
- Toprak O, Cirit M, Uzunel H, Ersoy R, Ermete M, et al. Hypocomplementaemic urticarial vasculitis syndrome and acute renal failure with cryoglobulin (–) hepatitis C infection. Nephrol Dial Transplant. 2004;19: 2680–2682. Ref.: https://goo.gl/UzcMfT
- Wisnieski JJ, Naff GB. Serum IgG antibodies to C1q in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. 1989; 32: 1119–1127. Ref.: https://goo.gl/XsWyHz
- Habif TP. Urticaria, angioedema, and pruritis. In: Clinical Dermatology. Elsevier Inc; 2015; 178–217.
- Fortson JS, Zone JJ, Hammond ME, Groggel GC. Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J Am Acad Dermatol. 1986; 15: 1137–1142. Ref.: https://goo.gl/s2iQgf
- Worm M, Sterry W, Kolde G. Mycophenolate mofetil is effective for maintenance therapy of hypocomplementaemic urticarial vasculitis. Br J Dermatol. 2000; 143: 1324. Ref.: https://goo.gl/a7DGGF
- Davis MD, Daoud MS, Kirby B, Gibson LE, Rogers RS 3rd. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol. 1998; 38: 899–905. Ref.: https://goo.gl/gyGcEM
- Trendelenburg M1, Courvoisier S, Späth PJ, Moll S, Mihatsch M, et al. Hypocomplementemic urticarial vasculitis or systemic lupus erythematosus? Am J Kidney Dis. 1999; 34: 745–751. Ref.: https://goo.gl/9xT68A