Case Report
Hypocomplementemic interstitial nephritis with long-term follow-up
Alyssa Penning, M.D1, Claire Kassakian, M.D2, Donald C Houghton, M.D1 and Nicole K Andeen, M.D1*
1Oregon Health & Science University, Department of Pathology, Portland, Oregon, USA
2Northwest Renal Clinic, Nephrology, Portland, Oregon, USA
*Address for Correspondence: Nicole Andeen, M.D, Oregon Health & Science University, Department of Pathology, 3181 SW Sam Jackson Park Road, Mail Code: L113, OR 97239, Portland, Oregon, USA, Tel: 503-494-8276; Fax: 503-494-2025; Email: [email protected]
Dates: Submitted: 17 January 2019; Approved: 22 February 2019; Published: 22 February 2019
How to cite this article: Penning A, Kassakian C, Houghton DC, Andeen NK. Hypocomplementemic interstitial nephritis with long-term follow-up. J Clini Nephrol. 2019; 3: 042-045. DOI: 10.29328/journal.jcn.1001024
Copyright License: © 2019 Penning A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Abstract
Prednisone-sensitive hypocomplementemia, renal insufficiency, and kidney biopsy demonstrating severe tubulointerstitial nephritis (TIN), storiform fibrosis, and tubulointerstitial immune deposits are typical of IgG4-related tubulointerstitial nephritis and hypocomplementemic interstitial nephritis. A diagnosis of hypocomplementemic interstitial nephritis requires clinical and pathologic exclusion of IgG4-related tubulointerstitial nephritis. We describe a patient with hypocomplementemic interstitial nephritis who did not develop diagnostic features of IgG4 related disease (RD) over 2-year follow-up. We conclude that hypocomplementemic interstitial nephritis could be on a biologic spectrum with IgG4-related disease, but not all cases will develop the abundance of IgG4-positive plasma cells, systemic manifestations, or elevated immunoglobulin levels characteristic of IgG4-RD.
Introduction
Idiopathic hypocomplementemic tubulointerstitial nephritis is characterized by hypocomplementemia and a robust interstitial nephritis with tubulointerstitial immune deposits [1]. It was described before the recognition of IgG4-TIN and is thought to be largely supplanted by this entity, due to the clinical and morphologic overlap. Many cases of hypocomplementemic interstitial nephritis likely represent IgG4-TIN, and it is unclear whether a biologically distinct entity of hypocomplementemic interstitial nephritis exists [2-5]. To the best of our knowledge, there is only one other reported case of hypocomplementemic interstitial nephritis in which systemic IgG4-RD, as currently defined, was explicitly excluded by histologic, clinical, and radiographic assessments at the time of biopsy [6].
Case Presentation
The patient is a 65-year-old man with a history of nephrolithiasis, mild right hydronephrosis, and non-steroidal anti-inflammatory use who presented with a creatinine of 1.7mg/dL without significant proteinuria or hematuria. He had no history of autoimmune disease, and infectious workup was negative. Laboratory studies revealed positive anti-nuclear antibody (ANA) at 1:80, negative anti-double-stranded DNA, and low complement levels. Serum protein electrophoresis (SPEP) demonstrated a polyclonal gammopathy consistent with chronic inflammation and/or infection; immunofixation and urine protein electrophoresis (UPEP) were negative for a monoclonal protein. Kappa and lambda light chains were elevated with a normal ratio. A kidney biopsy demonstrated severe tubulointerstitial nephritis with tubular basement membrane (TBM) immune deposits, without significant IgG4-positive plasma cells. He was treated with prednisone which was tapered and stopped after approximately 5 months; complement levels normalized and creatinine was at baseline of 1.7mg/dl. Four months later, his creatinine rose to 2.02mg/dl, complement levels dropped (C3: 57mg/dl, C4: <8mg/dl), and a repeat renal biopsy was performed.
Kidney biopsy demonstrated severe tubulointerstitial nephritis with abundant plasma cells, neutrophils, and eosinophils in a background of expansile and focally storiform interstitial fibrosis and tubular dropout (Figure 1). Immunofluorescence microscopy revealed TBM and interstitial immune deposits composed of polytypic IgG, C3, and C1q; IgG subclasses showed prominent staining for IgG1 and IgG3, with a lesser degree of IgG2 and IgG4. Deposits had dim staining for IgM without significant staining for IgA. There was focally moderately increased IgG4 positive plasma cells (generally 2/high powered field (hpf) but focally up to 12 /hpf), which comprised only a small portion (<5%) of CD138-positive plasma cells. Plasma cells were polyclonal by kappa and lambda in situ hybridization. Glomeruli appeared ischemic but without additional abnormalities. The prior biopsy had qualitatively similar findings with fewer tubulointerstitial immune deposits and IgG4-positive plasma cells (focally 1/hpf).
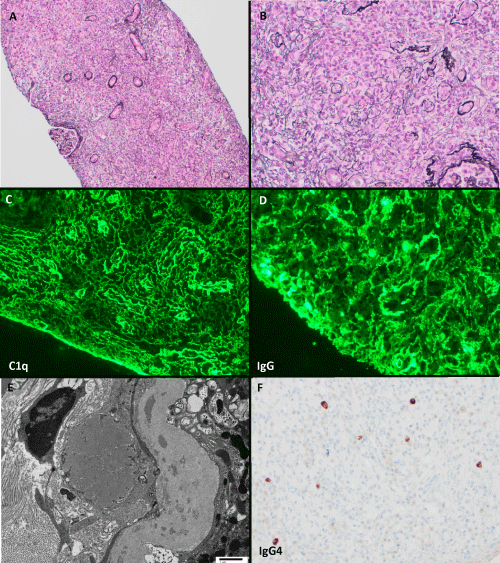
Figure 1: Edematous interstitium with tubular dropout, focally storiform fibrosis, and abundant plasma cells and eosinophils; glomeruli are compressed and ischemic (Jones stains, A: 100x, B: 200x). Interstitial and tubular basement membrane immune deposits are highlighted by C1q (C) and IgG (D). E) Ultrastructural studies demonstrate tubular basement membrane and interstitial immune deposits (transmission electron microscopy, direct magnification 4800x). F) IgG4 stain highlights focally increased IgG4 positive plasma cells (200x).
The patient was otherwise healthy with no systemic symptoms. At the time of the second biopsy, he had no recent therapy and total serum IgG and IgG4 levels were normal (IgG4: 8; reference range 1-123mg/dl). CT scan of the neck, chest, abdomen and pelvis showed no mass lesions. He had bilateral renal atrophy and mild mediastinal and hilar lymphadenopathy, considered to represent a reactive process or less likely a lymphoproliferative disorder. Repeat SPEP and UPEP were negative for a monoclonal protein. He received a second course of prednisone (3 months, shortened due to intolerance), and complement levels normalized and creatinine returned to baseline; these were stable for the following 7 months. Serum total IgG levels while off treatment remained normal. He developed hypocomplementemia a third time, which responded to low-dose prednisone and Rituximab (Table 1).
| Table 1: Summary of clinical and pathologic features. | |||
| At diagnosis | Repeat kidney biopsy at 9 months | Follow-up at 2 years | |
| Clinical and laboratory findings | |||
| Serum creatinine | 1.7 mg/dl | 2.02 mg/dl | 1.7 mg/dl |
| Proteinuria | No significant | No significant | No significant |
| ANA | Positive (1:80) | ||
| Anti-dsDNA | Negative | ||
| SPEP/UPEP and IFE | Polyclonal gammopathy consistent with chronic inflammation and/or infection. Negative immunofixation | Negative | |
| Serum complement levels | Low | Low (C3: 57mg/dl, C4: <8mg/dl) | Normal |
| Serum IgG & IgG4 levels | Normal (IgG4: 8, reference range 1-123mg/dl) |
Normal (Total IgG: 828, reference range 767-1590 mg/dl) |
|
| Imaging | CT chest, abdomen and pelvis with no mass lesions. Bilateral renal atrophy and mild lymphadenopathy | ||
| Subsequent treatment | Prednisone | Prednisone + Rituximab | None |
| Kidney biopsy | |||
| Light microscopy | Severe tubulointerstitial nephritis | Severe tubulointerstitial nephritis | |
| Tubular atrophy and interstitial fibrosis | Severe, focally storiform | Severe, focally storiform | |
| IgG4+ interstitial plasma cells | Generally 0, focally up to 1/hpf | Generally 2, focally up to 12/hpf | |
| %IgG4+ cells of total plasma cells | <5% | ||
| Immuno-fluorescence microscopy | Tubulointerstitial immune deposits+IgG, C3, C1q, k, l | Tubulointerstitial immune deposits+IgG, C3, C1q, k, l | |
| Electron microscopy | Tubular basement membrane immune deposits | Tubular basement membrane immune deposits | |
| ANA: Anti-nuclear antibody. Anti -dsDNA: anti-double stranded DNA. SPEP: serum protein electrophoresis. UPEP: Urine protein electrophoresis. IFE: immunofixation. | |||
Discussion
This case highlights the clinical and pathologic overlap of IgG4-TIN and hypocomplementemic TIN. The pathologic differential diagnosis of immune complex-mediated TIN with granular staining of tubular basement membranes also includes lupus nephritis (rare without glomerular involvement), Sjögren syndrome, anti-LDL receptor-related protein 2 nephropathy, membranous nephropathy, drug-induced TIN [7], subsequent to cardiac valve replacement and with associated giant cells [8], and infection, particularly polyoma virus nephropathy in the renal allograft [9]. Additional biopsy and clinical findings further narrow diagnostic considerations. Clinically, prednisone-sensitive hypocomplementemia with renal insufficiency are typical of IgG4-TIN or hypocomplementemic TIN.
Characteristic morphologic findings in IgG4-TIN include expansile storiform interstitial fibrosis, tubular basement membrane immune deposits, plasma-cell rich inflammation, and increased IgG4-positive interstitial plasma cells [10]. A cutoff of at least 10 IgG4-positive plasma cells/hpf is suggested [10], but this is not specific for IgG4-TIN and a modest increase in IgG4-positive interstitial plasma cells is seen in other renal diseases [11]. Proposed integrated criteria for systemic IgG4 related disease are characteristic histology plus serologic manifestations (elevated total IgG or IgG4) or clinical or radiographic evidence of other organ involvement [10,12,13]. Eighty-three percent of people with IgG4-TIN have other organ involvement, and 79% have an elevated serum total IgG or IgG4 [10]. In this patient, neither diagnostic serologic nor radiographic features of IgG4-RD were present, nor did they develop over two years of observation. We acknowledge that development of these systemic could have been blunted by therapy, but less than a third of the follow-up time was on disease-altering therapy, and serum immunoglobulin levels and imaging studies were normal at time of biopsy when the disease was active. Morphologically, both the essential absence of IgG4-positive plasma cells from the first kidney biopsy and the only modest increase in second kidney biopsy are uncharacteristic for IgG4-TIN. Thus this patient did not develop convincing clinical, pathologic, or radiographic criteria for a diagnosis of IgG4-TIN at diagnosis, on repeat biopsy, or during 2 years of follow-up.
As our understanding of the biology and temporal evolution of IgG4-RD improves, it is plausible that this and other cases of hypocomplementemic interstitial nephritis may be best considered in the IgG4-RD spectrum, representing incompletely understood cases with shared biologic underpinnings but which have not met criteria IgG4-RD. In this patient, however, repeat kidney biopsies and 2-year observation do not provide specific evidence that hypocomplementemic interstitial nephritis will develop the abundance of IgG4-positive plasma cells, systemic manifestations, or elevated immunoglobulin levels characteristic of IgG4-RD.
References
- Kambham N, Markowitz GS, Tanji N, Mansukhani MM, Orazi A, et al. Idiopathic hypocomplementemic interstitial nephritis with extensive tubulointerstitial deposits. Am J Kidney Dis. 2001; 37: 388-399. Ref.: https://goo.gl/NBKxhM
- Cornell LD. IgG4-related kidney disease. Diagnostic Histopathology. 2013; 19: 166-174. Ref.: https://goo.gl/w6QrEa
- Jeong HJ, Shin SJ, Lim BJ. Overview of IgG4-Related Tubulointerstitial Nephritis and Its Mimickers. J Pathol Transl Med. 2016; 50: 26-36. Ref.: https://goo.gl/ZEesDz
- Sprangers B, Claes K. IgG4-related disease should be considered in cases of hypocomplementemic immune-complex tubulointerstitial nephritis. NDT Plus. 2010; 3: 326. Ref.: https://goo.gl/n9dRd9
- Vaseemuddin M, Schwartz MM, Dunea G, Kraus MA. Idiopathic hypocomplementemic immune-complex-mediated tubulointerstitial nephritis. Nat Clin Pract Nephrol. 2007; 3: 50-58. Ref.: https://goo.gl/AyZ3VQ
- Kidder D, Stewart GA, Furrie E, Fleming S. The case. Idiopathic hypocomplementemic interstitial nephritis. Diagnosis: Idiopathic hypocomplementemic tubulointerstitial nephritis. Kidney Int. 2015; 87: 485-486. Ref.: https://goo.gl/VoihcQ
- Dixit MP, Nguyen C, Carson T, Guedes B, Dixit NM, et al. Non-steroidal anti-inflammatory drugs-associated acute interstitial nephritis with granular tubular basement membrane deposits. Pediatr Nephrol. 2008; 23: 145-148. Ref.: https://goo.gl/ChpTMa
- Chang A, Peutz-Kootstra CJ, Kowalewska J, Logar CM, Gitomer JJ, et al. Giant cell tubulitis with tubular basement membrane immune deposits: a report of two cases after cardiac valve replacement surgery. Clin J Am Soc Nephrol. 2006; 1: 920-924. Ref.: https://goo.gl/ti7bd2
- Bracamonte E, Leca N, Smith KD, Nicosia RF, Nickeleit V, et al. Tubular basement membrane immune deposits in association with BK polyomavirus nephropathy. Am J Transplant. 2007; 7: 1552-1560. Ref.: https://goo.gl/b9oCdH
- Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011; 22: 1343-1352. Ref.: https://goo.gl/9Ackuy
- Houghton DC, Troxell ML. An abundance of IgG4+ plasma cells is not specific for IgG4-related tubulointerstitial nephritis. Mod Pathol. 2011; 24: 1480-1487. Ref.: https://goo.gl/Bqb8gw
- Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012; 25: 1181-1192. Ref.: https://goo.gl/bhWAad
- Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011; 15: 615-626. Ref.: https://goo.gl/GU8itC