
More Information
Submitted: 01 January 2020 | Approved: 17 January 2020 | Published: 20 January 2020
How to cite this article: Darwich NS, Schnell M, Cossey LN. Granulomatosis with polyangiitis (GPA) in a 76-year old woman presenting with pulmonary nodule and accelerating acute kidney injury. J Clini Nephrol. 2020; 4: 001-006.
DOI: 10.29328/journal.jcn.1001048
Copyright License: © 2020 Darwich NS, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: GPA; ANCA-associated Vasculitides (AAV); Wegener granulomatosis

Granulomatosis with polyangiitis (GPA) in a 76-year old woman presenting with pulmonary nodule and accelerating acute kidney injury
Noor Sameh Darwich1*, Melissa Schnell2 and L. Nicholas Cossey3
1Miami Valley Hospital, Dayton, OH 45409, USA
2Department of Medicine, Miami Valley Hospital, Dayton, OH 45409, USA
3Arkana Laboratories, Little Rock, Arkansas, 72211, USA
*Address for Correspondence: Noor Sameh Darwich, MD. Miami valley Hospital, One Wyoming Street, Dayton, OH 45409, USA, Email: nsdarwich@gmail.com
Granulomatosis with polyangiitis (GPA), a form of ANCA-associated vasculitis (AAV), is a rare disease with an often-occult presentation. It is more common in 4th and 5th decades of life but can be seen in all ages.
This case report details a 76-year-old female presenting with abdominal pain, generalized weakness, and malaise, who was found to have pulmonary nodules on chest imaging. Biopsy of the lung nodule showed organizing pneumonia. Initially, antibiotics were used to treat the patient. However, she developed acute renal failure a few days after presentation and found to have positive serum C-ANCA as well as elevated ANCA-PR3 serologies. A subsequent kidney biopsy demonstrated pauci-immune necrotizing and crescentic glomerulonephritis that was consistent with GPA and the patient was started immediately on combination immunosuppressive therapy, plasmapheresis, and hemodialysis.
GPA’s clinical and radiological presentation can mimic other common conditions such as pneumonia, malignancy, bacterial sinusitis, pulmonary tuberculosis, sarcoidosis, and urinary tract infection. Because of this, a high level of suspicion is required for early diagnosis and treatment to alter the high mortality rate in this disease entity. All forms of ANCA-associated vasculitis (AAV) should be in the differential diagnosis for all patients presenting with multiorgan system involvement particularly in individuals with pulmonary and renal manifesations.
Antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides (AAV) is a spectrum of autoimmune diseases that include granulomatosis with polyangiitis (GPA) [formerly known as Wegener’s granulomatosis] [1], microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EPGA) [formerly known as Churg-Strauss syndrome] [2]. These conditions are associated with positive ANCA serologies and share similar pathological features on kidney biopsy including small vessel necrosis and, necrotizing and crescentic pauci-immune glomerulonephritis [3]. The clinical presentation and symptoms of GPA are not specific, and the disease can affect all organs. However, upper and lower respiratory tracts, lungs, kidney, and skin are the organs most commonly involved showing variable manifestations [4]. The most common presentation of GPA includes chronic rhinosinusitis not responding to conventional treatment, epistaxis, cough, fever, weight loss, fatigue, arthralgia, hemoptysis, dyspnea, hoarseness, oral ulcerations and oropharyngeal dysphagia [5]. Renal involvement is common in GPA and may initially present as mild microscopic hematuria. However, active urine sediment, gross hematuria, red blood cell cast formation, and accelerated acute kidney injury can also be seen. Diagnosis of GPA is often delayed because symptoms are nonspecific and may mimic other common diseases. Limited GPA disease usually affects upper and lower respiratory tracts or skin and is usually not initially life-threatening. However, GPA may progress to involve vital organs like the kidneys and lungs. Additionally, generalized severe GPA disease can occur with multisystem involvement and fatal outcome if not diagnosed and treated early before the disease advances.
Elevation of ANCA-PR3 is highly specific for GPA, and the sensitivity of this test is approximately 90% for patients with active disease. Current treatment options include combination immunosuppressive therapy [6]. Plasma exchange therapy (PET) has been widely used in patients with GPA presenting with severe progressive active renal disease or with pulmonary hemorrhage (7). However, a recent trial questioned the benefit of PET in the management of GPA. Hemodialysis is used when renal function deteriorates rapidly.
A 76-year-old female presented to the emergency department complaining of abdominal pain for the last two weeks which had progressively gotten worse, generalized weakness and malaise. She had a past medical history of chronic abdominal pain, gastroesophageal reflux disease and irritable bowel syndrome, asbestos exposure in the past, supraventricular tachycardia, and status post breast augmentation. She also described a dry non-productive cough. Her medications included lorazepam, lisinopril, buspirone, and aspirin. Her vital signs included: BP 115/58 mmHg, pulse 98 per minute, temp 98.3°F (36.8°C), respirations 16 per minute, and oxygen sat 96% on room air. Physical exam was noteworthy for moderate tenderness in the right upper quadrant and normal breath sounds bilateral. Laboratory data shown in table 1, was significant for a large amount of blood in the urine by dipstick urinalysis, CT abdomen and pelvis showed mild diverticulosis and increased stool in the colon. CXR demonstrated right diaphragm elevation (Figure 1).
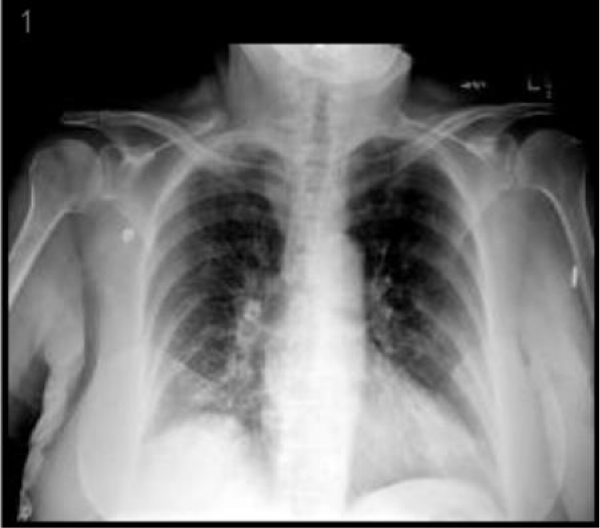
Figure 1: CXR demonstrated right diaphragm elevation.
Hepatobiliary iminodiacetic acid scan (HIDA scan) was negative for acute cholecystitis. Ultrasound of the abdomen was negative. A CT chest showed a Right upper lobe 2.9 cm pulmonary nodule and a right lower lobe 1.2 cm pulmonary nodule (Figures 2,3).
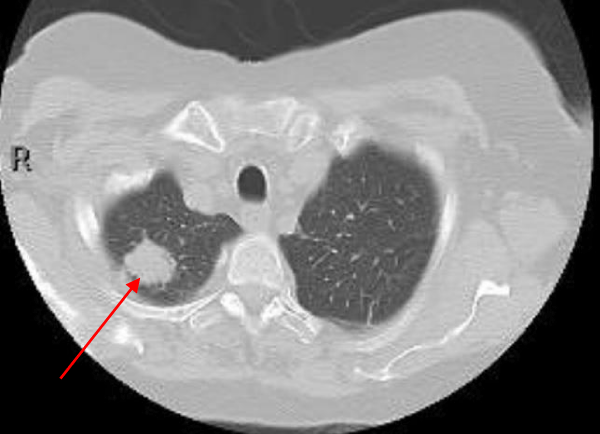
Figure 2: A CT chest showed a right upper lobe 2.9 cm pulmonary nodule.
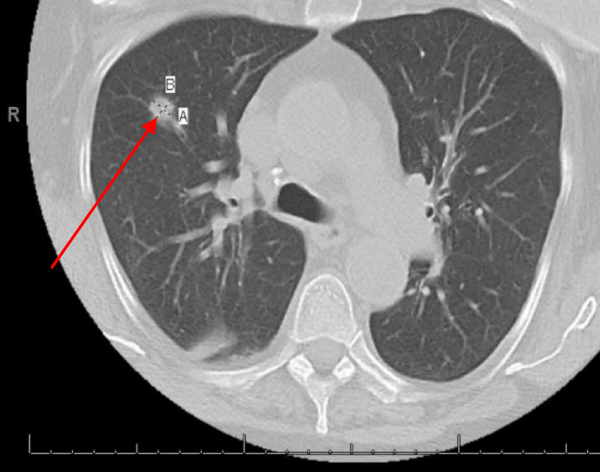
Figure 3: A CT chest showed right lower lobe 1.2 cm pulmonary nodule.
Positron Emission Tomography (PET scan) showed the right upper lobe pulmonary nodule with a Standardized uptake value (SUV) of 11.6 highly suspicious for malignancy (normal reference range < 2.5) (Figure 4).
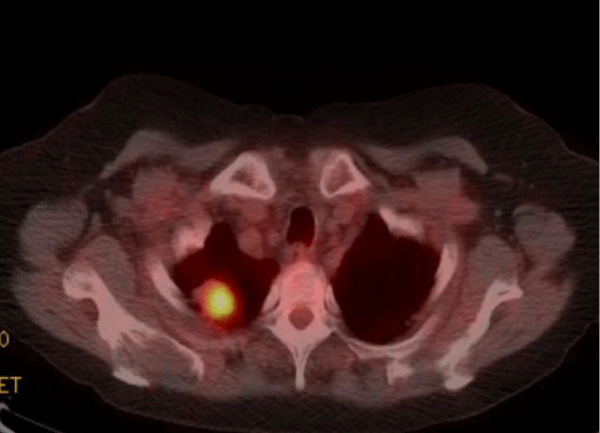
Figure 4: Positron Emission Tomography (PET scan) showed the right upper lobe pulmonary nodule with a Standardized uptake value (SUV) of 11.6 highly suspicious for malignancy (normal reference range <2.5).
The patient underwent CT guided lung biopsy of the right upper lobe nodule which showed acute organizing pneumonia with fibrosis, zonal necrosis but no granuloma and was negative for malignancy. Stains for adenovirus, aspergillus, cytomegalovirus, HSV1, HSV2, acid-fast and pneumocystis were all negative (Figures 5,6).
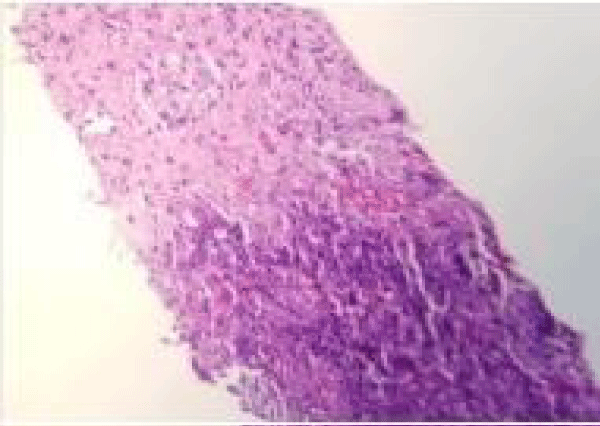
Figure 5: CT-guided lung biopsy of the right upper lobe nodule showed acute organizing pneumonia with fibrosis and zonal necrosis.
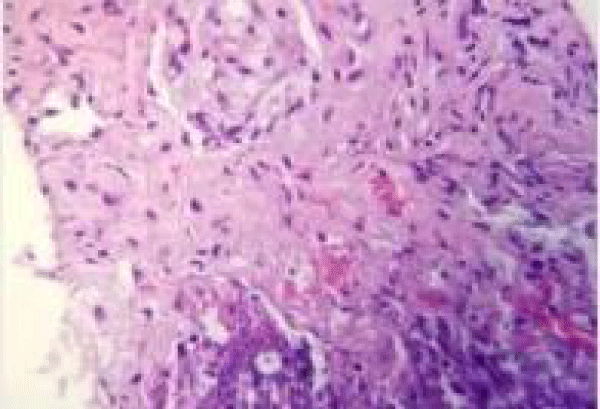
Figure 6: CT-guided lung biopsy of the right upper lobe nodule showed acute organizing pneumonia with fibrosis and zonal necrosis.
Her abdominal pain improved after treating her constipation with a bowel regimen. She was started on levofloxacin for a presumed diagnosis of pneumonia and was discharged home.
The patient presented again to the emergency department one week after her discharge complaining of right flank pain, weakness and bilateral lower extremity swelling, decrease appetite, soreness of left lateral edge of her tongue with odynophagia, diminished urine output, and persistent cough occasionally productive of yellowish sputum. On physical exam, she was found to have ulceration of the left lateral edge of her tongue and +1 pitting edema of the lower extremities. Her vital signs were BP 141/66 mmHg, pulse 116 per minute, temp 98,2°F (36.8°C), respirations 22 per minute. Laboratory test results shown in table 1, were significant for BUN 90 mg/dl, creatinine 10.60 mg/dl. Microscopic urinalysis was significant for +3 blood and red blood cells 50-75. Additionally, C-ANCA was positive with 1:320 titer and ANCA- PR3 IgG Antibody (autoantibodies to proteinase-3) positive with 205 EIA unit. However, anti-glomerular basement membrane antibodies, antinuclear (ANA) antibodies, and MPO IgG were all negative. The patient underwent ultrasound-guided left kidney biopsy which showed pauci-immune necrotizing and crescentic glomerulonephritis on a background of mild glomerular and tubulointerstitial chronicity (Figures 7,8).
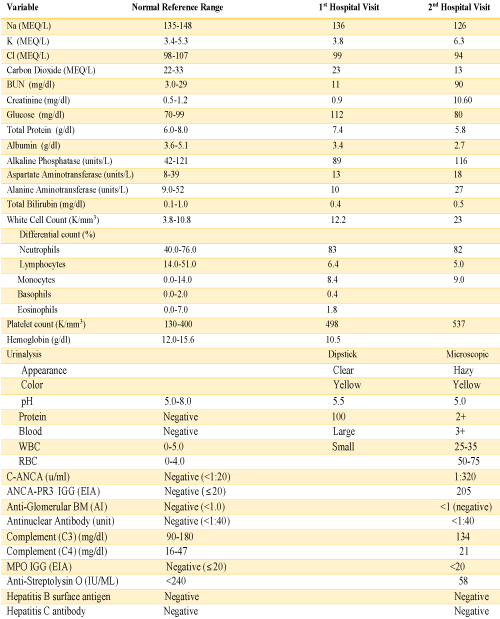
Table 1: Laboratory Data.
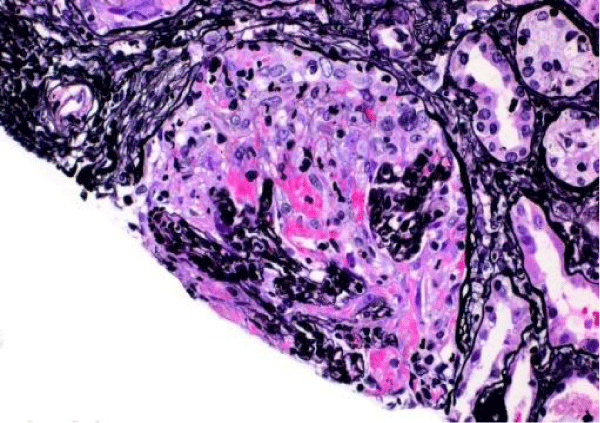
Figure 7: Global cellular crescent formation on JMS stain (200x) with glomerular tuft compression and a small area of fibrinoid necrosis (bright pink areas).
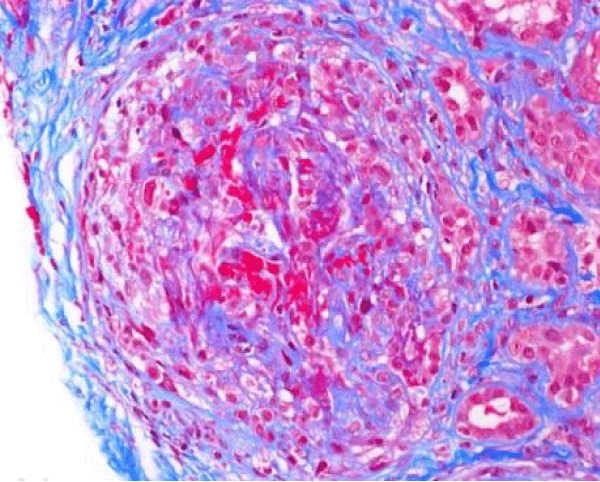
Figure 8: Global cellular crescent formation on Trichrome stain (200x) with central glomerular tuft obliteration by fibrinoid necrosis.
She was diagnosed with GPA and started on cyclophosphamide-glucocorticoids combination regimen with prophylactic trimethoprim-sulfamethoxazole (TMP/SMX) in addition to plasma exchange and hemodialysis.
ANCA-associated vasculitis (AAV) is a rare spectrum of diseases occurring predominantly in Caucasian people affecting both women and men equally in their 4th and 5th decades and is rarely reported in children and adolescence. The average incidence of GPA in the United States is 3 cases per 100,000 people annually with 90% of patients with GPA being Caucasian. GPA is a disease with unknown etiology but with genetic predisposition and environmental precipitating factors such as infection, environment allergens/irritants, exposure to solvents or silica, and variation in immune response to certain antigens. It can start insidiously with non-specific symptoms that can last for weeks or months before a diagnosis is made. Symptoms include fever, fatigue, weight loss, cough, hoarseness, stridor, wheezing, hearing loss, arthralgia, epistaxis, hemoptysis, dyspnea, dysphagia, abdominal pain, and pleuritic chest pain. The presentation can mimic common diseases such as pneumonia, malignancies, pulmonary tuberculosis, sarcoidosis, and viral, bacterial or fungal infections. Patient with GPA can present and progress rapidly over days or slowly over weeks and months [7].
GPA may affect a single organ, or it may involve several organs. It can involve almost all organ systems but the most common are upper and lower respiratory tracts which are seen in up to 90% of the patients including chronic rhinosinusitis, oral and nasal ulcers, otitis media, purulent/bloody nasal discharge, polychondritis with bone and cartilage destruction resulting in a saddle nose deformity and tracheal or subglottic stenosis [9].
Lung involvement in GPA is seen in the form of lung nodules or masses, sometimes with cavitation, pleural-based lesions, and patchy, diffuse infiltrates suggesting diffuse alveolar hemorrhage presenting as hemoptysis.
Renal involvement, which is seen in 80% of patients diagnosed with GPA, can be seen as the first presentation or later during the course of the disease and manifests initially by asymptomatic hematuria with active urine sediment, and rise in serum creatinine, proteinuria, and eventually acute kidney injury.
Skin manifestations of GPA can be in the form of purpura involving lower extremities, livedo reticularis, nodules, and ulcerations. Ophthalmic involvement by GPA is less common but typically presents as scleritis/episcleritis, which presents with eye redness and pain, conjunctivitis and corneal ulceration. More rare ophthalmic presentation of GPA includes Optic neuropathy, uveitis, retinal vasculitis with visual disturbances, diplopia, foreign body sensation in the eye and proptosis have also been reported [10]. Oral ulcers or gingivitis (strawberry gingivitis) have been seen in a patient with GPA [11]. Occasionally the gastrointestinal tract can be involved causing abdominal pain due to peritonitis, splanchnic vasculitis, and occasionally gastrointestinal bleeding [12,13]. Oropharyngeal dysphagia and odynophagia, as well as esophageal involvement, have been described in patients with GPA [14].
Up to 94% of patients with active generalized GPA have a positive cytoplasmic pattern of antineutrophilic cytoplasmic antibody (C-ANCA) primarily PR3-ANCA, while MPA is associated with MPO-ANCA. However, 10% of patients with GPA are ANCA negative by conventional serologic assays, and in patients with limited GPA disease who are presenting with an only upper or lower respiratory tract or skin involvement, C-ANCA may be positive in only 60% [15]. One explanation for these ANCA negative patients is that an epitope-masking factor in sera interferes with available serologic testing. Indeed, total ANCA titer does not correlate well with disease activity, and researchers have identified ANCA epitopes specific to active disease while other epitopes can persist during remission and some epitopes can even exist naturally in disease-free individuals suggesting that epitope specificity defines pathogenicity [16].
For a definitive diagnosis, a biopsy from the site of suspected active disease is needed. Lung or kidney is usually the site where the biopsy is done offering the highest diagnostic yield. However, other sites can be biopsied such as the skin and nasal site but with a much lower yield of diagnosis and limited value.
Tissue diagnosis can be elusive when using transbronchial biopsy which is often non-diagnostic requiring thoracoscopic or open lung biopsy [17]. Typically, lung biopsy shows necrotizing granulomas with small vessel vasculitis when the disease is active. However, granuloma associated with necrosis take time to develop, and If a diagnosis is made early during the disease course, granulomas may not be present.
Renal biopsy usually shows necrotizing and crescentic, pauci-immune glomerulonephritis and/or necrotizing vasculitis. The mortality rate is as high as 90% in 2 years if not recognized and treated early in the disease. If the biopsy cannot be done or needs to be delayed due to patient factors, then the diagnosis can be made with high probability based on clinical findings and serological testing with positive serum ANCA-PR3. Treatment should not be delayed pending tissue diagnosis as GPA can be fatal if not diagnosed and treated quickly. Combination immunotherapy treatment should be started in patients suspected of having a GPA with supporting clinical findings and positive serology for C-ANCA while the patient is being stabilized and prepared for biopsy. Most physicians will start with only high dose pulse intravenous steroids therapy pending confirmation of tissue diagnoses, and some will add oral cyclophosphamide to intravenous steroids while tissue biopsy is being undertaken and processed.
Current standard treatment of patients with GPA, as stated by most recent recommendations from the Kidney Disease: Improving Global Outcomes (KDIGO) [18], and the European league Against rheumatisom (EULAR) in conjuction with the European renal Association -European Dialysis and transplant Association (ERA-EDTA) [19], includes induction of remission by combination immunosuppressive therapy with high dose Glucocorticoids and either cyclophosphamide or rituximab, which are both equally effective in achieving remission in up to 75% of patient, followed by maintenance therapy for prolonged but variable periods of time and up to 2 years in some patients, with usually a single immunosuppressive agent such as azathioprine or Rituximab to maintain disease activity in remission with the goal of decreasing the incidence and severity of relapsing GPA [20,21]. Mycophenolate mofetile (MMF) and methotrexate has also been used but less commonly in maintenance therapy of patient with GPA. Rituximab, which is a B-cell depleting monoclonal antibody, an inhibitor of antibody production, is the first FDA approved medication for the treatment of ANCA-associated vasculitis including GPA and MP [22]. While Cyclophosphamide has been long used in patient with GPA, it is not FDA approved for the treatment of ANCA-associated vasculitis. However, some practitioners still prefer to use cyclophosphamide-based regimen for induction of remission therapy in selected patients with severe GPA while others shifting to a rituximab-based regimen for most patients [23]. Rituximab is considered the preferred and first line immunosuppressive agent for patients with GPA who had a relapsing disease, failed initial remission with cyclophosphamide-base regimen, or in whom cyclophosphamide is contraindicated [24]. Methotrexate in combination with glucocorticoids can be used as an initial induction therapy in patients with GPA presenting with rhinosinusitis, pulmonary nodules or arthritis with no evidence of organ-threatening or life-threatening disease. Renal replacement therapy with hemodialysis is indicated in patients with rapidly progressive worsening renal function. Currently, plasma exchange, as an adjunctive therapy, is usually added to immunosuppresive therapy in severe cases of GPA with rapidly declining Glomerular filtration rate (GFR) including a serum creatinine above 4.0 mg/dl or who require hemodialysis and for patients presenting with diffuse pulmonary hemorrhage associated with respiratory failure [25]. However, a recent data from PEXIVAS trial, Plasma Exchange and Glucocorticoids for Treatment of Anti-Neutrophil Cytoplasm Antibody (ANCA)- Associated Vasculitis, which has not yet published, questioned the utility of plasma exchange in patient with ANCA-associated vasculitis as it did not significantly reduced mortality nor avoided the development of end stage renal disease when Plasma exchange used as an adjuvant modality with immunosuppressive therapy [26]. Key factors which determine mortality and outcome in patients with GPA are early recognition and treatment. With the currently available immunotherapy, more than 50% of patients with GPA recover renal function and become dialysis independent and 5 years survival rate is up to 80%. After treatment with immunosuppressive therapy, most morbidity in GPA patients is treatment related.
Up to 50% of patients with GPA in remission may relapse within 5 years of diagnosis. Risk factors for relapse include persistence of PR3-ANCA positivity despite clinical remission, history of upper airway diseases such as sinusitis and subglottic stenosis, or lung involvement [27]. Nasal carriage of Staphylococcus aureus is associated with GPA flare-ups and relapse, and the use of trimethoprim-sulfamethoxazole (TMP-SMX) as prophylaxis can reduce the incidence of relapse in patients with GPA on the basis of eradication of bacteria and stopping the initiating antigen stimulus [28]. TMP-SMX is also used in patients with GPA started on immunotherapy as prophylaxis for PCP [29].
Diagnosis of granulomatosis with polyangiitis (GPA) can be challenging and the consequences detrimental when misdiagnosis occurs. GPA is a rare disease with low physician awareness but should be kept in the differential diagnosis of those patients who presents with non-specific symptoms. Marked heterogeneity of presentation can occur and include hematuria, hemoptysis, chronic rhinosinusitis, recurrent urinary tract infection and multisystem presentation especially when upper or lower respiratory tract involvement is present. When strong clinical suspicion for GPA exists, early diagnosis and initiation of disease arresting immunosuppressive therapy are suggested as progression can occur quickly and can be potentially irreversible. Serologies can be misleading in a minority of patients and treatment modalities vary depending on the underlying extent and severity of the disease [30]. Furthure research aimed at better understanding the pathogenesis of GPA and the development of new novel more effective agent with less side effect is warranteed. Avacopan, which is a selective complemet (C5a) receptor inhibitor is being investigated as a glucocorticoid- sparing agent in the management of GPA with promising result [31,32].
- Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DR, et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011; 63: 863-864. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21374588
- Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitis. Am J Med. 2004; 117: 39-50. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15210387
- Gomez-Puerta JA, Hernandez-Rodriguez J, Lopez-Soto A, Bosch X. Antineutrophil cytoplasmic antibody-associated vasculitides and respiratory disease. Chest 2009; 136: 1101-1111. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19809051
- Jayne D. The diagnosis of vasculitis. Best Pract Res Clin Rheumatol.. 2009; 23: 445-453. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19508950
- Sedighi S, Aghaie M, Mozafari N, Roshandel G. Different features in Wegener’s Granulomatosis: report of five cases. Journal of Clinical and Diagnostic Research. 2011(Suppl-2), 5:1437-1439.
- Specks U, Merkel PA, Seo P, Spiera R, Langford CA, et al. Efficacy of remission-induction regimens for ANCA- associated vasculitis. N Eng J Med. 2013; 369: 417-427. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23902481
- Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, et al. Randomized trial of plasma exchange or High-dose methylprednisolone A adjunctive therapy for sever renal vasculitis. J Am Soc Nephrol. 2007; 18: 2180-218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17582159
- Langford CA, Hoffman GS. Wegener’s granulomatosis. Thorax 1999; 54: 629-637. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1745525/
- Shafiei K, Luther E, Archie M, Gulick J, Fowler MR. Wegener’s granulomatosis: Case report and brief literature review. J Am Board Fam Pract. 2003; 16: 555-559. PubMed:
- Sfiniadaki E, Tsiara I, Theodossiadis P, Chatziralli I. Ocular manifestations of Granulomatosis polyangiitis: A review of the Literature. Ophthalmol Ther (2019) 8:227-234. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14963084
- Stewart C, Cohen D, Bhattacharyya I, Scheitler L, Riley S, et al. Oral manifestations of Wegener’s granulomatosis: a report of three cases and literature review. J Am Dent Assoc. 2007; 138: 338-348. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17332039
- Storesund B, Gran JT, Koldingsnes W. Severe intestinal involvement in wegener’s granulomatosis: report of two cases and review of the literature. Br J Rheumatol. 1998; 37: 387-390. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9619888
- Pagnoux C, Mahr A, Cohen P, Guillevin L. Presentation and outcome of gastrointestinal involvement in systemic necrotizing vasculitidies: analysis of 62 patients with polyarteritis nodosa, microscopic polyangiitis, Wegener granulomatosis, Churg-Strauss syndrome, or rheumatoid arthritis-associated vasculitis. Medicine (Baltimore). 2005; 84: 115128. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15758841
- Miller PG, Santini C, Freed MJ. Dysphagia in a patient with Wegener's Granulomatosis: a Case report. Dysphagia. 2001; 16: 136-139. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11305224
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, et al. 2012 revised international chapel Hill onsensus conference Nomenclature of vasculitides. Arthritis Rheum. 2013; 65: 1-11. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23045170
- Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest. 2013; 123: 1773-1783. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23549081
- Schnabel A, Holl-Ulrich K, Dalhoff K, Reuter M, Gross WL. Efficacy of transbronchial biopsy in pulmonary vasculitidies. Eur Respir J. 1997; 10: 2738-2743. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9493653
- Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, et al. Management and treatment of glomerular Diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. 2019; 95: 281-295; PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30665569
- Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, et al. EULAR/ERA-EDTA recommendations for the Management of ANCA-associated vasculitis. Ann Rheum Dis 2016; 75: 1583-1594. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27338776
- Tesar V, Rihova Z, Jancova E, Rysava R, Merta M, et al. Current treatment strategies in ANCA-positive renal vasculitis: lessons from European randomized trials. Nephrol Dial Transplant. 2003; 18: Suppl 5: V2-V4. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12817056
- Charles P, Néel A, Tieulié N, Hot A, Pugnet G, et al. Rituximab for induction and maintenance treatment of ANCA-Associated vasculitides: a multicenter retrospective study on 80 patients. Rheumatology. 2014; 53: 532-539. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24282319
- Getha D, Kallenberg C, Stone JH, Salama AD, Appel GB, et al. Current therapy of Granulomatosis with polyangiitis and microscopic Polyangiitis: the role of rituximab. J Nephrol. 2015; 28: 17-27. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25185728
- Merkel PA, Kaplan AA, Falk RJ, Granulomatosis with polyangiitis and microscopic Polyangiitis: intial immunosuppressive therapy. UpToDate. 2020; 1-37.
- Jones RB, Furuta S, Tervaert JW. Rituximab versus cyclophosphamide in ANCA-Associated renal vasculitis: 2-year results of a randomized trial. Ann Rheum Dis. 2015; 74: 1178-1182. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25739829
- Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, Henke DC, et al. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis. American Journal of Kidney Diseases 2003; 42: 1149-1153. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14655185
- Walsh M, Merkel PA, Jayne. The Effects of Plasma Exchange and reduced-Dose Glucocorticoids during Remission-Induction for Treatment of Sever ANCA-Associated Vasculitis [abstract]. Arthritis Rheumatol. 2018: 70 (Suppl 10). https://acrabstracts.org/abstract/the-effects-of-plasma-exchange-and-reduced-dose-glucocorticoids-during-remission-induction-for-treatement-of-sever-anca-associated-vasculitis/
- Hogan SL, Folk RJ, Chin H, Cai J, Jennette CE, et al. Predictors of relapse and treatment resistance in Antineurtophil Cytoplasmic Antibody-Associated Small-vessel Vasculitis. Ann Intern Med. 2005; 143: 621-631. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16263884
- Popa ER, Trevaert JW. The relation between Staphylococcus aureus and Wegener’s Granulomatosis: Current knowledge and future Directions. Intern Med. 2003; 42: 771-780. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14518661
- Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-Sulfamethoxazole(CO-TRIMOXAZOLE) for the prevention of relapses of Wegener’s granulomatosis. For the Dutch Co-Trimoxazole Wegener Study group. N Engl J Med. 1996; 335: 16-20. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8637536
- Geetha D, Jefferson JA. ANCA-Associated Vasculitis: Core Curriculum 2020. Am J Kidney Dis. 2020; 75: 124-137. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31358311
- Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, et al. CLEAR study Group. Randomized trial of C5a Receptor inhibitor Avocopan in ANCA-Associated Vasculitis. J AM Soc nephrol. 2017; 28: 2756-2767. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28400446
- Salvadori M, Tsalouchos A. Antineutrophil cytoplasmic antibody associated Vasculitides with renal involvement: Open challenges in the remission induction Therapy. World J nephrol. 2018; 7: 71-83. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29736379