
More Information
Submitted: June 26, 2023 | Approved: July 31, 2023 | Published: August 01, 2023
How to cite this article: Nessa L, Singh A, Sharif MW, Enabi J, Bashir M. Diagnostic Challenge of Gitelman Syndrome: A Rare but Significant Cause of Electrolyte Imbalance. J Clini Nephrol. 2023; 7: 053-056.
DOI: 10.29328/journal.jcn.1001109
Copyright License: © 2023 Nessa L, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Diagnostic Challenge of Gitelman Syndrome: A Rare but Significant Cause of Electrolyte Imbalance
Lutfor Nessa1*, Arjan Singh1, Muhammad Waqar Sharif1, Joud Enabi1 and Mamoun Bashir2
1Resident Physician, Department of Internal Medicine, Texas Tech University Health Sciences Center, Permian Basin, USA
2Nephrologist, Texas Tech University Health Sciences Center, Permian Basin, USA
*Address for Correspondence: Lutfor Nessa, Resident Physician, Department of Internal Medicine, Texas Tech University Health Sciences Center, Permian Basin, USA, Email: lunessa@ttuhsc.edu
Objective: This case study presents a young female patient diagnosed with symptomatic electrolyte disturbances, later confirmed as Gitelman syndrome (GS). It highlights the underlying pathophysiology and emphasizes the importance of its proper management.
Background: GS is a rare genetic disorder affecting kidney electrolyte reabsorption, leading to symptoms like weakness, muscle cramps, fatigue, nausea, and vomiting. Diagnosis involves lab tests and genetic confirmation, with treatment comprising electrolyte supplementation and medications. Ongoing management is vital to prevent complications.
Case presentation: A 23-year-old Caucasian female presented to the ED with sudden weakness in all extremities, thirst, and lightheadedness. Lab results showed hyperglycemia 166 (70-100 mg/dL), severe hypokalemia 1.1 (3.6-5.1 mmol/L), mild hypercalcemia 11 (8.9-10.4 mg/dL), and severe hypophosphatemia 0.6 (2.3-7.0 mg/dL). Incidentally, she had prior hypokalemia history from a motor accident hospitalization and managed it with KCl for a year but stopped when symptoms improved. She was treated with electrolyte replacement and discharged with oral potassium. Five days later, she returned with severe hypokalemia 1.3, mild hypercalcemia 10.7, and severe hypophosphatemia 0.6. A 24-hour urinary test showed distal convoluted tubulopathy indicative of GS. She was treated with replacement therapy and spironolactone, with instructions for ongoing supplementation and follow-up with a nephrologist.
Discussion: GS is mostly caused by mutations in the SLC12A3 gene, affecting the kidneys’ sodium chloride cotransporter function, as confirmed in our patient.
Conclusion: While GS has no cure, appropriate treatment with medication and dietary adjustments can enhance patients’ quality of life by maintaining electrolyte balance. Healthcare providers’ awareness is crucial for effective care and complication prevention.
Gitelman syndrome (GS) is an autosomal recessive disorder that affects the kidneys’ ability to reabsorb electrolytes, including potassium and magnesium [1]. It also presents with metabolic alkalosis, hypocalciuria, and hyperreninemic hyperaldosteronism [2]. This salt-losing tubulopathy is also referred to as familial hypokalemia-hypomagnesemia. The prevalence of GS is approximately 1 in 40,000 people, with an estimated 1% of individuals being heterozygous carriers [3]. However, the exact prevalence remains unknown, though it tends to be more prevalent in the Asian population [3].
GS is associated with various symptoms of hypokalemia and hypomagnesemia, such as muscle weakness, tetany, fatigue, nausea, vomiting, and palpitations [4]. While rare, sudden cardiac deaths have been reported in some cases. However, GS may present as biochemical alterations with or without specific manifestations, making timely recognition vital, especially in young patients with critical serum potassium values. A descriptive study of five GS patients revealed that laboratory findings played a significant role in diagnosis, with malaise being the only acknowledged clinical symptom [5]. The mean potassium was 2.5 ± 0.5 mmol/l, and the serum magnesium value was 1.3 ± 0.3 mg/dl
[5]. Diagnosing GS can be challenging due to its rarity, necessitating a high level of clinical suspicion, a thorough analysis of serum and urine profiles, along with genetic testing, to achieve a definitive diagnosis [6].
Patients with GS require careful monitoring for the development of extrarenal complications, such as short stature, febrile convulsions, thyroid dysfunction, epilepsy, or QT interval (QT) prolongation [6]. Interestingly, it has been observed that certain variants of SLC12A3 may not cause severe hypomagnesemia [6].
Treatment usually involves increasing electrolyte intake, using drugs to aid electrolyte retention by the kidneys, and, in severe cases, IV electrolyte replacement therapy [8]. Additionally, a low-sodium diet and potassium-sparing diuretics may be beneficial for managing symptoms [9]. In this case study, a 23-year-old female was diagnosed with Gitelman syndrome five years after the initial presentation.
A 23-year-old Caucasian female with a history of recurrent hypokalemia presented to the Emergency Department (ED) with sudden onset of generalized weakness that started in the upper extremities and progressed to the lower extremities, leaving her unable to walk without assistance. She reported receiving an unknown injection at an urgent care clinic for a sinus infection and taking one dose of Cefdinir before the onset of symptoms. The patient also complained of excessive thirst and lightheadedness but denied other gastrointestinal or cardiovascular symptoms. On arrival, vital signs were as follows: blood pressure 110/60 mmHg, heart rate 104/min, respiratory rate 18/min, and temperature 36.7 degrees Celsius. Physical examination was normal except for an obese body habitus, dry oral mucosa, and strength in both upper extremities rated 4/5 and both lower extremities rated 3/5. Electrocardiogram (EKG) showed a prolonged QT of 767 ms and T wave inversions. Laboratory investigations showed hyperglycemia (166 mg/dL), severe hypokalemia (1.1 mmol/L), mild hypercalcemia (11 mg/dL), and severe hypophosphatemia (0.6 mg/dL).
The patient had a history of hypokalemia, which was incidentally found during a hospitalization for a motor vehicle accident five years prior. She had previously been managed with KCl 20 mEq oral daily for a year but stopped taking the medication as her symptoms improved. She occasionally took an unknown over-the-counter pill but was unsure of its name, content, and dosage. The patient was adopted, so family history was limited, but according to the family, her biological maternal uncle had hypokalemia.
The patient was admitted to the ICU, and a nephrologist was consulted. She was started on intravenous potassium phosphate (KPO4) 30 mmol every 6 hours and oral KPO4 40 mmol every 4 hours, along with Normal Saline (NS) at a rate of 200 cc/hr. A day after admission, the hypophosphatemia resolved, so the treatment was changed to oral potassium chloride (KCL) 40 meq every 4 hours, intravenous KCL 30 meq every 6 hours, and 0.45% NS with 40 meq of KCL at 100 cc/hr. Gradually, her potassium level and Corrected QT Interval (QTc) improved to 3.6 mmol/L and 460 ms, respectively. She was discharged with oral potassium supplementation of 20 meq twice daily and instructions to follow up with the nephrologist for further evaluation and management of her hypokalemia.
Five days later, the patient returned to the hospital with weakness. Although she had been feeling well after being discharged and returning to work, she had been experiencing flu-like symptoms since her previous admission and visited an outside ED, where she received an injection and was advised to continue taking cefdinir. Shortly after receiving the injection, she began to experience weakness and fatigue, the same symptoms that led to her previous admission. The patient had been compliant with KCl 20 mEq oral twice daily, but her hypokalemia had worsened, reaching severe levels (K: 1.3 mmol/L). The patient denied recent changes in diet, gastrointestinal symptoms, or medication alterations.
Vitals were normal, and physical examinations revealed an obese body habitus with somnolence, upper extremity strength rated 3/5 and lower extremity strength rated 5/5. Laboratory investigations showed hyperglycemia (189 mg/dL), severe hypokalemia (1.3 mmol/L), mild hypercalcemia (10.7 mg/dL), and severe hypophosphatemia (0.6 mg/dL).
The patient was managed in the ICU, and potassium was replaced through a central line for faster and easier administration. She received KCL 80 meq IV and 40 meq orally in the ED. In the ICU, she was started on NS at 125 cc/hr along with potassium supplementation using oral KCL 40 meq every 4 hours and IV KCL 20 meq every 2 hours. An EKG on admission showed a prolonged QTc of 606, which improved by the following day. A nephrologist was consulted again, and a 24-hour urinary test indicated distal convoluted tubulopathy, consistent with Gitelman syndrome, showing potassium, magnesium, sodium loss, and calcium retention. The patient was treated with potassium and magnesium replacement therapy and spironolactone. Upon discharge, she was instructed to continue potassium and magnesium supplementation and spironolactone and to follow up with a nephrologist. At the time of discharge, her potassium level returned to her baseline of 3.3 mmol/L, QTc improved to 423 ms, and she was safely discharged home. Subsequent genetic testing confirmed the diagnosis, showing two heterozygous variants in the SLC12A3 gene.
Table 1 displays the Complete Blood Count (CBC) and glucose levels for the previous admission and readmission.
| Table 1: Hematology. | ||||||
| Test Name | Units | Reference Range |
Previous Admission |
Discharge | Readmission | Discharge |
| WBC | 10*3/uL | 4.8-10.8 | 8.8 | 9.5 | 8.1 | 11.0 |
| RBC | 10*3/uL | 4.0-5.2 | 5.2 | 4.5 | 4.7 | 4.6 |
| Hgb | g/dL | 12-16 | 14.9 | 13.2 | 13.7 | 13.3 |
| Platelet | 10*3/uL | 150-400 | 262 | 209 | 264 | 202 |
| Glucose | mg/dL | 70-100 | 166 | 85 | 189 | 92 |
| WBC: White Blood Cell Count; RBC: Red Blood Cell Count; Hgb: Hemoglobin; Platelet: Platelet Count; Glucose: Blood Sugar level. | ||||||
Table 2 compares the basic metabolic panel of the previous admission and readmission.
| Table 2: Biochemistry. | ||||||
| Test Name | Units | Reference Range |
Previous Admission |
Discharge | Readmission | Discharge |
| Sodium | mmol/L | 136-144 | 140 | 138 | 143 | 138 |
| Potassium | mmol/L | 3.6-5.1 | 1.1 | 3.6 | 1.3 | 3.3 |
| Chloride | mmol/L | 96-106 | 102 | 102 | 105 | 99 |
| Bicarb | mmol/L | 21-31 | 23 | 25 | 20 | 24 |
| Anion Gap | mmol/L | 6-16 | 14.9 | 11 | 17.7 | 12 |
| Calcium | mg/dL | 8.9-10.4 | 11 | 9.4 | 10.7 | 9.0 |
| Magnesium | mg/dL | 1.8-2.5 | 1.8 | 1.8 | 2.0 | 2.0 |
| Phosphorus | mg/dL | 2.3-7.0 | 0.6 | 3.9 | 0.6 | 5.2 |
Table 3 shows hormone levels on admission, with Aldosterone and Renin levels being within the normal range during the first admission.
| Table 3: Hormone Levels on Admission. | |||
| Test Name | Units | Reference Range | On Admission |
| Aldosterone | ng/dL | Upright: 4.0 - 31.0 Supine: Less than or equal to 16.0 Unspecified: Less than or equal to 31.0 |
12.8 |
| Renin | ng/mL/hr |
Adult, Normal sodium diet: Supine: 0.2-1.6 Upright: 0.5-4.0 |
5.4 |
Table 4 presents 24-hour urine creatinine and electrolyte measurements for both previous admission and readmission.
| Table 4: 24-hour Urine Creatinine and Electrolytes. | ||||
| Test Name | Units | Reference Range |
Previous Admission |
Readmission |
| 24-hour urine creatinine | mg/day | 1000-1800 | 1018 | 1118 |
| 24-hour urine sodium | mmol/day | 40-220 | 743 | 235 |
| 24-hour urine potassium | mmol/day | 25-150 | 27 | 230 |
| 24-hour urine magnesium | mg/day | 73-122 | 243 | 142 |
| 24-hour urine calcium | mg/day | 100-240 | < 20 | N/A |
Hypokalemia is a prevalent finding in hospitalized patients, estimated to affect 20% of cases [10]. Most causes of hypokalemia are related to intracellular shift, increased excretion, and poor intake. Although most of the time the causes can be delineated, at times it is difficult to find a specific cause, especially in patients with persistent hypokalemia when other causes have been ruled out.
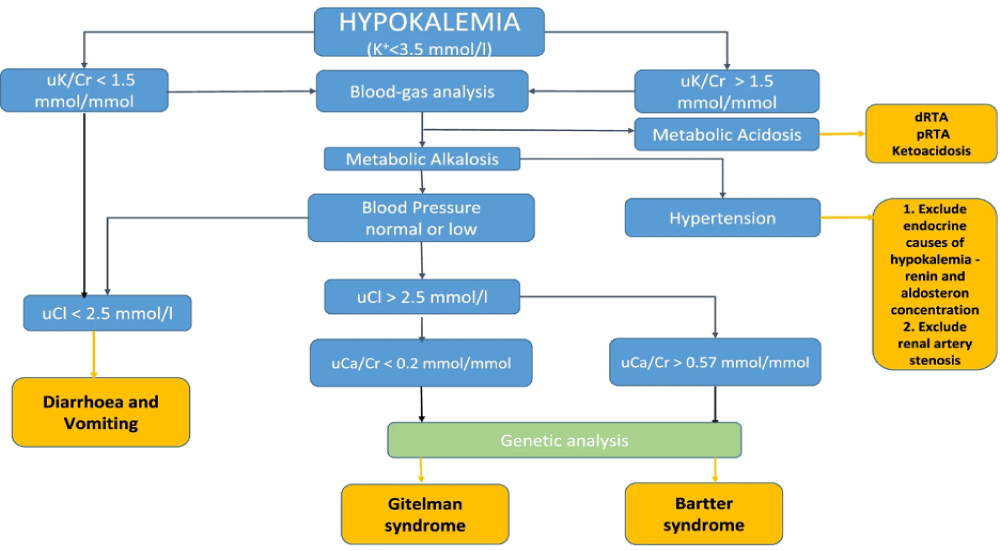
GS is an autosomal recessive renal tubulopathy usually presenting during adolescence or adulthood (Parmar, et al. 2022). It is a salt-losing tubulopathy arising from an inactivating mutation in the thiazidesensitive Na-Cl cotransporter gene, SLC12A3 [11,12], and is usually overlooked given its rare occurrence. It is characterized by renal loss of potassium, magnesium, and sodium and retention of calcium, mimicking thiazide diuretic effects. Hence patients tend to have metabolic alkalosis with hypokalemia, hypomagnesemia, and hypocalciuria [12].
In our case, the patient was a young lady with a history of recurrent hypokalemic events manifesting as muscle weakness progressing to paralysis and QT prolongation in EKG. Given her prolonged course and multiple prior admissions, we tried ruling out as many differentials as possible. The patient had no other comorbidities, had a normal thyroid function, and denied the use of diuretics or insulin. Moreover, she had a good appetite without any gastrointestinal symptoms like nausea, vomiting, or diarrhea. Therefore, gastrointestinal loss and poor oral intake were not considered.
With this, our differential shifted towards renal etiology and subsequently, the 24-hour urinary analysis showed elevated potassium (230 mmol/day) and magnesium (142 mg/day) levels with reduced urine calcium level(< 20 mg/day). This was consistent with distal convoluted tubulopathy.
The patient was euglycemic and normotensive, and serum aldosterone and renin were normal. Hence renal potassium-losing etiologies such as primary aldosteronism, Cushing syndrome, renin tumor, and rare conditions like Liddle syndrome were ruled out. Renal tubular acidosis was not considered as the patient’s laboratory findings were suggestive of metabolic alkalosis. Bartter syndrome is another rare tubulopathy that arises from mutations in genes that encode transporters and channels responsible for salt reabsorption in the thick ascending limb [13]. It is known to cause normal or increased urinary calcium as opposed to hypocalciuria seen in GS [14].
Importantly, in GS, the loss-of-function mutations in SLC12A3 impair NCC-mediated Na+ reabsorption, and similar phenotypes can arise from mutations in other genes, including CLCKNB, KCNJ10, FXYD2, HNF1B, MT-TI, MT-TF, KCNJ16, and ATP1A1, emphasizing the role of cell metabolism and basolateral membrane potential in Na+ reabsorption [15]. Therefore, genetic testing should cover a panel of genes associated with Gitelman-like syndromes, including mitochondrial genes [15]. However, in our case, genetic testing confirmed the presence of mutations in the SLC12A3 gene, confirming the diagnosis of Gitelman syndrome.
In this case, we try to focus on how easily GS can be misdiagnosed or even missed. It is necessary to consider the possibility of GS in patients with recurrent clinical hypokalemia in the setting of hypomagnesemia and hypocalciuria.
In conclusion, GS requires prompt diagnosis and management to address electrolyte imbalances. While there is no cure for GS, proper treatment with medications and dietary adjustments can help maintain stable electrolyte levels. Potassium and magnesium supplements are commonly prescribed to replenish depleted levels, and potassium-sparing diuretics may also be used to prevent further losses. Dietary changes, such as incorporating a potassium and magnesium-rich diet, play a vital role in managing the condition. Regular follow-ups with healthcare professionals are essential to tailor treatment plans to individual needs. Early recognition of symptoms and seeking medical attention promptly can prevent complications and ensure better outcomes. Proper management empowers patients to lead fulfilling lives and improves their overall well-being, allowing them to effectively manage the challenges associated with GS.
Ethical approval: This research did not involve the use of any patient identifiers; therefore, ethical approval was not required for this study.
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221-35. PMID: 5929460.
- Parmar MS, Muppidi V, Bashir K. Gitelman Syndrome. In StatPearls. StatPearls Publishing. 2023.
- Kondo A, Nagano C, Ishiko S, Omori T, Aoto Y, Rossanti R, Sakakibara N, Horinouchi T, Yamamura T, Nagai S, Okada E, Shima Y, Nakanishi K, Ninchoji T, Kaito H, Takeda H, Nagase H, Morisada N, Iijima K, Nozu K. Examination of the predicted prevalence of Gitelman syndrome by ethnicity based on genome databases. Sci Rep. 2021 Aug 9;11(1):16099. doi: 10.1038/s41598-021-95521-6. PMID: 34373523; PMCID: PMC8352941.
- Urwin S, Willows J, Sayer JA. The challenges of diagnosis and management of Gitelman syndrome. Clin Endocrinol (Oxf). 2020 Jan;92(1):3-10. doi: 10.1111/cen.14104. Epub 2019 Oct 6. PMID: 31578736.
- Ungaro CM, Odstrcil-Bobillo MS, Russo PM. Síndrome de Gitelman [Gitelman syndrome]. Medicina (B Aires). 2020;80(1):87-90. Spanish. PMID: 32044746.
- Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, Ellison DH, Karet Frankl FE, Knoers NV, Konrad M, Lin SH, Vargas-Poussou R. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017 Jan;91(1):24-33. doi: 10.1016/j.kint.2016.09.046. PMID: 28003083.
- Fujimura J, Nozu K, Yamamura T, Minamikawa S, Nakanishi K, Horinouchi T, Nagano C, Sakakibara N, Nakanishi K, Shima Y, Miyako K, Nozu Y, Morisada N, Nagase H, Ninchoji T, Kaito H, Iijima K. Clinical and Genetic Characteristics in Patients With Gitelman Syndrome. Kidney Int Rep. 2018 Sep 28;4(1):119-125. doi: 10.1016/j.ekir.2018.09.015. PMID: 30596175; PMCID: PMC6308995.
- Bakir M, Ibrahim HAG. A Challenging Case of Persisting Hypokalemia Secondary to Gitelman Syndrome. Cureus. 2021 Oct 10;13(10):e18636. doi: 10.7759/cureus.18636. PMID: 34765380; PMCID: PMC8576546.
- Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008 Jul 30;3:22. doi: 10.1186/1750-1172-3-22. PMID: 18667063; PMCID: PMC2518128.
- Kardalas E, Paschou SA, Anagnostis P, Muscogiuri G, Siasos G, Vryonidou A. Hypokalemia: a clinical update. Endocr Connect. 2018 Apr;7(4):R135-R146. doi: 10.1530/EC-18-0109. Epub 2018 Mar 14. PMID: 29540487; PMCID: PMC5881435.
- Galli-Tsinopoulou A, Patseadou M, Hatzidimitriou A, Kokka P, Emmanouilidou E, Lin SH, Tramma D. Gitelman syndrome: first report of genetically established diagnosis in Greece. Hippokratia. 2010 Jan;14(1):42-4. PMID: 20411059; PMCID: PMC2843570.
- Chen SY, Jie N. Gitelman syndrome: A case report. World J Clin Cases. 2022 Jun 16;10(17):5893-5898. doi: 10.12998/wjcc.v10.i17.5893. PMID: 35979117; PMCID: PMC9258353.
- Cunha TDS, Heilberg IP. Bartter syndrome: causes, diagnosis, and treatment. Int J Nephrol Renovasc Dis. 2018 Nov 9;11:291-301. doi: 10.2147/IJNRD.S155397. PMID: 30519073; PMCID: PMC6233707.
- Fulchiero R, Seo-Mayer P. Bartter Syndrome and Gitelman Syndrome. Pediatr Clin North Am. 2019 Feb;66(1):121-134. doi: 10.1016/j.pcl.2018.08.010. PMID: 30454738.
- Schlingmann KP, de Baaij JHF. The genetic spectrum of Gitelman(-like) syndromes. Curr Opin Nephrol Hypertens. 2022 Sep 1;31(5):508-515. doi: 10.1097/MNH.0000000000000818. Epub 2022 Jul 11. PMID: 35894287; PMCID: PMC9415222.