
More Information
Submitted: November 02, 2023 | Approved: November 11, 2023 | Published: November 14, 2023
How to cite this article: Prabhu H, Vellanki V, Dhake S, Karunanithi S. Rare Case of Dense Deposition Disease with Combined C3 and C4d Deposits with MYH9-related Mutation. J Clini Nephrol. 2023; 7: 097-101.
DOI: 10.29328/journal.jcn.1001117
Copyright License: © 2023 Prabhu H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: C3 glomerulopathy; Dense deposition disease; Drusen; Classical complement pathway; MYH9 nephropathy

Rare Case of Dense Deposition Disease with Combined C3 and C4d Deposits with MYH9-related Mutation
Harish Prabhu1*, Venkat Vellanki2, Suvarna Dhake3 and Sathiyan Karunanithi4
1Department of Nephrology, Burjeel Royal Hospital, Al Ain, Abu Dhabi, United Arab Emirates
2Department of Radiology, Burjeel Royal Hospital, Al Ain, Abu Dhabi, United Arab Emirates
3Department of Ophthalmology, Burjeel Royal Hospital, Al Ain, Abu Dhabi, United Arab Emirates
4Burjeel Royal Hospital, Al Ain, Abu Dhabi, United Arab Emirates
*Address for Correspondence: Harish Prabhu, Department of Nephrology, Burjeel Royal Hospital, Al Ain, Abu Dhabi, United Arab Emirates, Email: drharishprabhu@gmail.com
Background: The C3 glomerulopathies are a group of rare forms of glomerulonephritis with an incidence of 1-2 cases per million. It is mainly characterized by dysregulation of the alternative complement pathway. It is further classified morphologically based on electron microscopy ultrastructural findings into Dense Deposition Disease (DDD) and C3 glomerulonephritis. DDD is normally characterised by C3 Deposits.
Case: We report a rare case of a young Emirati male who presented with sub nephrotic proteinuria and microscopic haematuria on routine evaluation. Renal biopsy showed features of DDD with combined C3 and C4 deposits. The retinal evaluation showed features of Drusen classically seen in DDD. Genomic study showed heterozygous mutation in c.5842G>C (p.Asp1948His) variant of uncertain significance in MYH9 gene.
Discussion: C3 Glomerulopathy is a type of immune mediated disease previously classified as membranoproliferative glomerulonephritis. DDD is mainly characterised by C3 deposits in the glomerular basement Membrane. Our case has both C3 and C4d deposits, which is a rare entity. It shows the activation of both classical and alternate pathways.
Conclusion: Dense deposition disease is a rare complement mediated glomerulopathy. It is characterised by C3 deposits. Dense deposition disease with combined C3 and C4d deposits is a new entity. The treatment and prognosis of such cases will be different and unique compared to the normal cases of DDD.
The C3 glomerulopathies are a group of rare forms of glomerulonephritis with an incidence of 1-2 cases per million. It is a form of complement-mediated glomerulopathy characterized by dysregulation of the alternative complement pathway [1]. Light microscopic features can vary from near normal glomerulus to membrano proliferative pattern, crescents, endocapillary pattern,mesangioprolifertiave pattern etc. It is further classified morphologically based on electron microscopy ultrastructural findings into Dense Deposition Disease and C3 glomerulonephritis.
Dense Deposition Disease (DDD) was first described by Berger and Galle in 1962. The term DDD is used based on the characteristic appearance of linearappearing, highly electron-dense material in the glomerular basement membrane on an electron microscope. These deposits can also be seen sometimes in the mesangium, Bowman’s capsule, and tubular basement membrane. Historically dense deposition disease was classified as a subgroup of primary membranoproliferative glomerulonephritis (MPGN type 2).
Recently it was reclassified as a complement-mediated glomerular disease [2].
C3 Glomerulonephritis is characterised by isolated deposits of C3 on immunofluorescence but instead of dense intramembranous deposits as in DDD, electron microscopy reveals predominant subendothelial and mesangial electron-dense deposits of lesser intensity. Cases of DDD with C3 deposits have been reported and a few cases of C4 deposits have also been reported. We report a rare case of DDD with combined C3 and C4 deposits in a patient with MYH9 mutation. MYH9 nephropathy with DDD has never been reported before.
A 26-year-old, Emirati gentleman presented to the nephrology outpatient clinic for evaluation of proteinuria detected on routine evaluation as a part of fitness for work. On examination, he was moderately built and nourished with a blood pressure of 128/70 mmhg. His blood workup showed normal blood count and normal renal function with a creatinine of 64 umol/L. Urine showed 2 + protein and microscopic hematuria. 24hour urine protein showed a protein loss of 776 mg. Ultrasound abdomen showed normalsized kidneys with normal echotexture with preserved cortical medullary differentiation.
His glomerular disease workup was negative for ANA, anti-ds DNA, anti-MPO, and anti-PR3. The complement study showed low C3 0.78 g/dl (normal 0.9 -1.8 g/dl) and normal complement 4. Virology markers including hepatitis b, hepatitis C, and HIV were negative to diagnose his underlying renal pathology, he underwent a left percutaneous renal biopsy under ultrasound guidance. The biopsy specimen was subjected to light microscopy, immunofluorescence, and electron microscopy.
Light microscopy showed 14 glomeruli,1 of them sclerosed. The glomerular basement membrane showed PAS positivity and shows global duplication (Figure 1), but no spikes or holes in the silver stain. 5% tubular atrophy and interstitial scarring were noted. Interlobular arteries did not show any lesions.
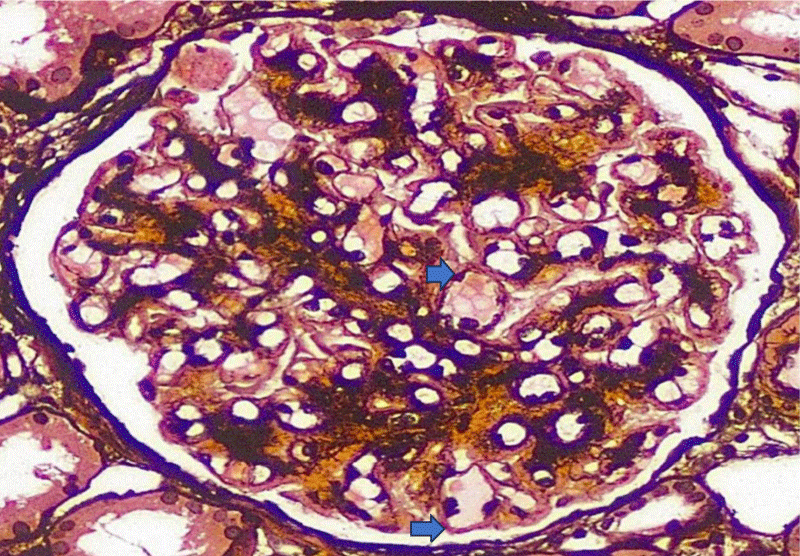
Figure 1: The glomerular basement membrane shows PAS positivity and global basement membrane duplication (blue arrows.
Immunofluorescence showed 8 perfused glomeruli. Frozen sections were stained for IgG, IgM, IgA, C3, C1q, C4d, kappa & lambda light chains, PLA2R, IgG4, THSDS7A, Albumin & Fibrinogen. There is interrupted GBM and mesangial positivity for IgM (traces) and C3. Surprisingly there was positive staining for C4d (2+) and C5b9 (2+).
Electron microscopy showed almost 100% of the podocyte foot processes are effaced. There was an extensive, interrupted, segmental deposition of intensely electron-dense material within the glomerular basement membrane replacing lamina densa (Figure 2). Similar material was seen in the mesangium and segmentally within the basement membranes of Bowman’s capsule and tubules. Mesangial matrix was increased. All the features were suggestive of Dense Deposition Disease.
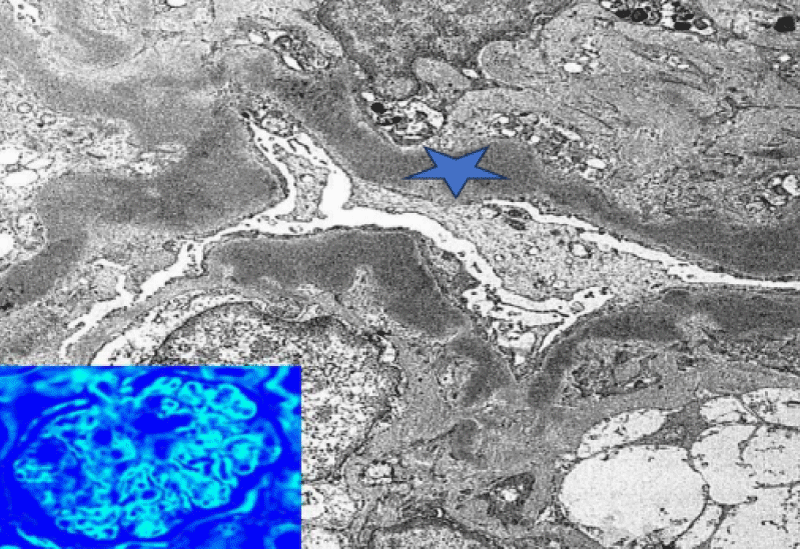
Figure 2: Electron microscope showing electron - dense material within the glomerular basement membrane (Blue star).
The retinal evaluation was done and showed features of Drusen (Figures 3-5) were present in both eyes, which is more classically seen in dense deposition disease than C3 glomerulonephritis.

Figure 3: Drusen appearing as small yellow deposits in the dilated eye examination (Blue arrow).
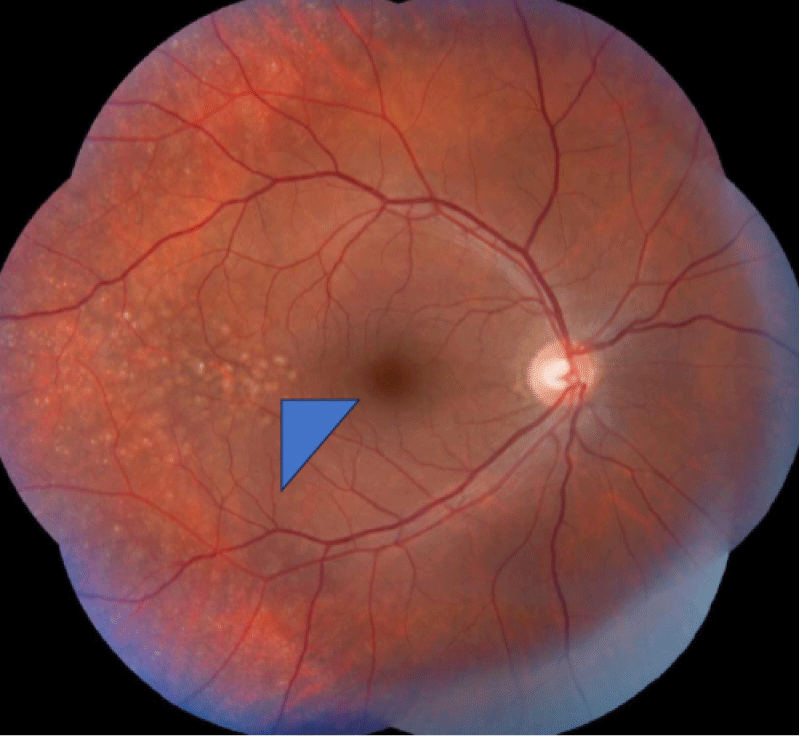
Figure 4: Digital Color fundus Examination showing small yellow spots on fundus representing drusen (Blue triangle head).
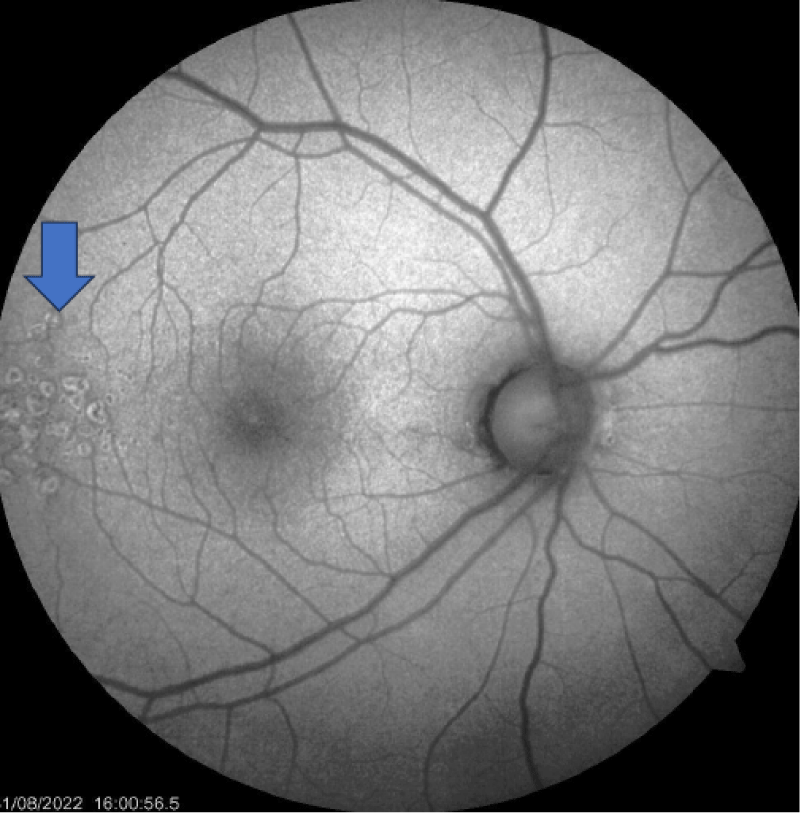
Figure 5: Monochromatic Fundus examination showing drusen (Blue arrow).
C3 nephritic factor (C3NeF) was sent as it is the most common autoantibody detected in dense deposition disease but it was tested negative.
Serum protein electrophoresis was done to rule out any plasma cell dyscrasias and it was also negative. Genetic study clinical exome sequencing was done and it showed the presence of a heterozygous mutation in c.5842G>C (p.Asp1948His) variant of uncertain significance in MYH9 gene.
C3 Glomerulopathy was first described in 2007 [3]. Over the years the disease has gained more importance with better diagnostic tools and low threshold for renal biopsies.
It was initially classified as membranoproliferative glomerulonephritis.
In 2013 it was classified as an independent Glomerulo-pathy in “2013 Consensus of the International Society of Nephrology” [4]. It was further classified into Dense Deposit Disease (DDD) and Complement 3 Glomerulonephritis (C3GN). Further classifications was based on the appearance and distribution of deposits on electron microscopy of the glomerulus.
DDD is characterized by the presence of highly electron-dense deposits within the glomerular basement membrane (GBM), while in C3GN the deposits are located in the mesangium or along the subendothelial side of the GBM [5] DDD is named based on the appearance of hypereosinophilic electron-dense deposits in the middle layer (lamina densa) of the Glomerular Basement Membrane (GBM). These deposits can be seen in the mesangium, bowman’s capsule, and tubular basement membrane.
The pathogenesis of both DDD and C3GN is thought to be due to excessive activation of the alternate complement pathway. Genetic alterations of regulatory and activation processes of alternate pathway results in excess activation of alternate pathway. It is mainly due to either increased activity of C3 convertase by C3 nephritic factor.
(C3NeF) an autoantibody stabilizing C3 convertase [6,7] or functional loss of factor H activity. In addition to genetic factors, acquired factors have also been found to have a role in the pathogenesis of C3G. Reports of antibodies against B factor, H factor and C5 convertase have been documented.
Genetic factors like homozygous Cfh deficiency, homozygous loss of function Cfh mutation, heterozygous gain of function C3 mutation and CFHR mutations leading to enhanced Cfh deregulation [8] have all been reported as causes for DDD. Regardless of genetic or acquired factors, the final C5 common complement pathway is activated leading to disease manifestations.
Clinical presentation of DDD includes acute nephritic syndrome (16% to 38%), isolated macroscopic hematuria, microscopic haematuria with sub nephrotic-range proteinuria, isolated proteinuria and nephrotic syndrome [9,10]. DDD has also been reported with an initial presentation of synphargitic haematuria [11]. It can affect both paediatric and adult populations. For adults above 50 years of age, monoclonal gammopathy should be ruled out [12].
Treatment of DDD is challenging as there no randomized clinical trials or clinical guidelines. All patients should be started on diet control, salt restriction, control of dyslipidaemia, control of blood pressure and initiation of ACE inhibitors or angiotensin I receptor blockers to inhibit the renin-angiotensin system and reduce urinary protein.
The prognosis from observational studies shows that more than 50% of patients with DDD of 10 or more years to progress to ESRD.
Patients with urinary protein of more than 500 mg in 24 hrs and moderate Inflammation on biopsy can be started on mycophenolate and steroids. Based on case series reports 66% of patients responded to MMF or steroid treatment. [13,14] Periodic plasma infusions have been found to be beneficial in patients with a defect in Factor H [15]. For patients with rapidly progressive renal failure, gluco-corticoids in combination with either cyclophosphamide or MMF should be considered.
Eculizumab has been found to be very effective in patients with refractory disease not responding to steroids and MMF. Evidence of eculizumab is from case reports and series and one open labelled clinical trial [16-18].
The prognosis of DDD is worse than C3GN. Overall 70% of children progress to CKD as shown in a study in which cases were followed for a period of 9 years [7]. Follow-up studies in adults showed that 50% will progress to ESKD [19]. Renal failure and age at time of presentation were the poor prognostic factors.
Our patient had normal renal functions with 24 hr urine protein of 776 mg at the time of presentation with microscopic heamturia. Renal biopsy light microscopy and electron microscopy showed features of DDD. It showed C3 deposits. Surprisingly our case also showed C4d deposits along with C3 deposits. Sethi, et al. have described 3 cases of C4d deposits. They are classified as CC4d glomerulopathy similar to C3 glomerulopathy. C4d glomerulopathy is further classified in to C4dGlomerunonephritis (C4GN) and C4d DDD following their observation of three cases [20]. Our case showed features of both C3 deposits and C4d deposits which have only been described once in literature before [21]. It raised the possibility of another entity of DDD, that is, combined C3 and C4d in DDD in addition to C3DDD and C4d DDD. Further studies and case reports are required for confirmation of the same. Our patient was started on angiotensin receptor blockers along with salt restriction and lifestyle modifications. The dose of ARBs was gradually increased to maximum tolerated dose. After 3 months his proteinuria has increased to more than 2 gm and was not responding to supportive treatment. He was started on steroids and myfortic acid. His renal functions are stable and he achieved partial remission with the latest proteinuria of 800 mg in 24 hrs.
Clinical exome genetic sequencing showed our patient
also had MYH9 gene-related heterozygous mutation. MYH9 gene-related nephropathy is a rare disease and has been reported before in few cases. 30% of cases of MYH9-related disease develop nephropathy. It is characterised by proteinuria and rapidly progressive renal failures. The previously reported cases of MYH9-related nephropathy have shown segmental glomerulosclerosis, focal global glomerulosclerosis, global glomerulosclerosis and foot process effacement [20,22-25].
Our Patient has diffuse foot process effacement. However, features of dense deposition disease in MYH9 nephropathy have never been reported before. However, our patient did not have any extrarenal features of MYH9 Nephropathy like giant platelets, thrombocytopenia, cataracts, and sensorineural deafness. Our case is the first case with DDD with MYH9 gene mutation having combined C3 and C4d deposits. The possibility MYH9 genetic mutation in the causation of dense deposition disease is less as he does not have any extra renal features of MYH9 Nephropathy. His parents also underwent genetic screening. Father also had heterozygous mutation in c.5842G>C (p.Asp1948His) variant of uncertain significance in the MYH9 gene. Father had mild proteinuria of 260 mg with normal renal functions and no hematuria. A kidney biopsy was not done for the father.
C3Glomerulopathy is a rare form of immune-mediated glomerulopathy characterised by C3 deposits. It is morpho-logically divided into dense deposition disease and C3 glomerulonephritis based on electron microscope findings.
In dense deposition disease, the deposits are predominantly due to the activation of alternate complement pathway. The presence of C4D deposits in our cases raises the possibility of an activation of the classical pathway. Combined C3 and C4D deposits raised the possibility of a new entity of Dense Deposition Disease with activation of both classical and alternate pathways.
The treatment and prognosis of such cases will also be different from the cases of DDD reported so far considering the rarity of the disease and different pathogenesis.
It will be interesting to see future cases of DDD with combined C3 and C4D deposits. More studies and case reports will give more clarity regarding the treatment options and prognosis.
Declarations
Informed consent was obtained from the Individual whose case is reported. The Case study was done in accordance with the World Medical Association Declaration of Helsinki.
- Smith RJH, Appel GB, Blom AM, Cook HT, D'Agati VD, Fakhouri F, Fremeaux-Bacchi V, Józsi M, Kavanagh D, Lambris JD, Noris M, Pickering MC, Remuzzi G, de Córdoba SR, Sethi S, Van der Vlag J, Zipfel PF, Nester CM. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol. 2019 Mar;15(3):129-143. doi: 10.1038/s41581-018-0107-2. PMID: 30692664; PMCID: PMC6876298.
- Walker PD, Ferrario F, Joh K, Bonsib SM. Dense deposit disease is not a membranoproliferative glomerulonephritis. Mod Pathol. 2007 Jun;20(6):605-16. doi: 10.1038/modpathol.3800773. Epub 2007 Mar 30. PMID: 17396142.
- Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, Grünfeld JP, Lesavre P, Noël LH, Fakhouri F. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007 Mar;44(3):193-9. doi: 10.1136/jmg.2006.045328. Epub 2006 Oct 3. PMID: 17018561; PMCID: PMC2598029.
- Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, Doyle M, Fakhouri F, Fervenza FC, Fogo AB, Frémeaux-Bacchi V, Gale DP, Goicoechea de Jorge E, Griffin G, Harris CL, Holers VM, Johnson S, Lavin PJ, Medjeral-Thomas N, Paul Morgan B, Nast CC, Noel LH, Peters DK, Rodríguez de Córdoba S, Servais A, Sethi S, Song WC, Tamburini P, Thurman JM, Zavros M, Cook HT. C3 glomerulopathy: consensus report. Kidney Int. 2013 Dec;84(6):1079-89. doi: 10.1038/ki.2013.377. Epub 2013 Oct 30. PMID: 24172683; PMCID: PMC3842953.
- Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, Doyle M, Fakhouri F, Fervenza FC, Fogo AB, Frémeaux-Bacchi V, Gale DP, Goicoechea de Jorge E, Griffin G, Harris CL, Holers VM, Johnson S, Lavin PJ, Medjeral-Thomas N, Paul Morgan B, Nast CC, Noel LH, Peters DK, Rodríguez de Córdoba S, Servais A, Sethi S, Song WC, Tamburini P, Thurman JM, Zavros M, Cook HT. C3 glomerulopathy: consensus report. Kidney Int. 2013 Dec;84(6):1079-89. doi: 10.1038/ki.2013.377. Epub 2013 Oct 30. PMID: 24172683; PMCID: PMC3842953.
- Schwertz R, Rother U, Anders D, Gretz N, Schärer K, Kirschfink M. Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: a long-term follow-up. Pediatr Allergy Immunol. 2001 Jun;12(3):166-72. doi: 10.1034/j.1399-3038.2001.012003166.x. PMID: 11473682.
- Schwertz R, de Jong R, Gretz N, Kirschfink M, Anders D, Schärer K. Outcome of idiopathic membranoproliferative glomerulonephritis in children. Arbeitsgemeinschaft Pädiatrische Nephrologie. Acta Paediatr. 1996 Mar;85(3):308-12. doi: 10.1111/j.1651-2227.1996.tb14022.x. PMID: 8695987.
- Barbour TD, Pickering MC, Terence Cook H. Dense deposit disease and C3 glomerulopathy. Semin Nephrol. 2013 Nov;33(6):493-507. doi: 10.1016/j.semnephrol.2013.08.002. PMID: 24161036; PMCID: PMC3820036.
- Sibley RK, Kim Y. Dense intramembranous deposit disease: new pathologic features. Kidney Int. 1984 Apr;25(4):660-70. doi: 10.1038/ki.1984.71. PMID: 6482170.
- Habib R, Gubler MC, Loirat C, Mäiz HB, Levy M. Dense deposit disease: a variant of membranoproliferative glomerulonephritis. Kidney Int. 1975 Apr;7(4):204-15. doi: 10.1038/ki.1975.32. PMID: 1095806.
- Bennett WM, Fassett RG, Walker RG, Fairley KF, d'Apice AJ, Kincaid-Smith P. Mesangiocapillary glomerulonephritis type II (dense-deposit disease): clinical features of progressive disease. Am J Kidney Dis. 1989 Jun;13(6):469-76. doi: 10.1016/s0272-6386(89)80004-6. PMID: 2658560.
- Ravindran A, Fervenza FC, Smith RJH, Sethi S. C3 glomerulopathy associated with monoclonal Ig is a distinct subtype. Kidney Int. 2018 Jul;94(1):178-186. doi: 10.1016/j.kint.2018.01.037. Epub 2018 May 3. Erratum in: Kidney Int. 2018 Nov;94(5):1025. PMID: 29729982; PMCID: PMC7735221.
- Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, Roccatello D, Cheung M, Wheeler DC, Winkelmayer WC, Floege J; Conference Participants. Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019 Feb;95(2):281-295. doi: 10.1016/j.kint.2018.11.008. PMID: 30665569.
- Avasare RS, Canetta PA, Bomback AS, Marasa M, Caliskan Y, Ozluk Y, Li Y, Gharavi AG, Appel GB. Mycophenolate Mofetil in Combination with Steroids for Treatment of C3 Glomerulopathy: A Case Series. Clin J Am Soc Nephrol. 2018 Mar 7;13(3):406-413. doi: 10.2215/CJN.09080817. Epub 2018 Jan 11. PMID: 29326307; PMCID: PMC5967675.
- Licht C, Heinen S, Józsi M, Löschmann I, Saunders RE, Perkins SJ, Waldherr R, Skerka C, Kirschfink M, Hoppe B, Zipfel PF. Deletion of Lys224 in regulatory domain 4 of Factor H reveals a novel pathomechanism for dense deposit disease (MPGN II). Kidney Int. 2006 Jul;70(1):42-50. doi: 10.1038/sj.ki.5000269. Epub 2006 Apr 12. PMID: 16612335.
- Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D'Agati VD, Canetta PA, Radhakrishnan J, Appel GB. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012 May;7(5):748-56. doi: 10.2215/CJN.12901211. Epub 2012 Mar 8. PMID: 22403278; PMCID: PMC3338285.
- Le Quintrec M, Lapeyraque AL, Lionet A, Sellier-Leclerc AL, Delmas Y, Baudouin V, Daugas E, Decramer S, Tricot L, Cailliez M, Dubot P, Servais A, Mourey-Epron C, Pourcine F, Loirat C, Frémeaux-Bacchi V, Fakhouri F. Patterns of Clinical Response to Eculizumab in Patients With C3 Glomerulopathy. Am J Kidney Dis. 2018 Jul;72(1):84-92. doi: 10.1053/j.ajkd.2017.11.019. Epub 2018 Feb 9. PMID: 29429752.
- Daina E, Noris M, Remuzzi G. Eculizumab in a patient with dense-deposit disease. N Engl J Med. 2012 Mar 22;366(12):1161-3. doi: 10.1056/NEJMc1112273. Erratum in: N Engl J Med. 2012 Apr 12;366(15):1454. PMID: 22435382.
- Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, Markowitz GS, D'Agati VD. Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol. 2009 Jan;4(1):22-32. doi: 10.2215/CJN.03480708. Epub 2008 Oct 29. PMID: 18971369; PMCID: PMC2615696.
- Sethi S, Quint PS, O'Seaghdha CM, Fervenza FC, Bijol V, Dorman A, Dasari S, Smith RJ, Kurtin PJ, Rennke HG. C4 Glomerulopathy: A Disease Entity Associated With C4d Deposition. Am J Kidney Dis. 2016 Jun;67(6):949-53. doi: 10.1053/j.ajkd.2016.01.012. Epub 2016 Feb 17. PMID: 26896898.
- Vankalakunti M, Augustine R, Jangamani R, Siddini V, Bonu R, Babu K, Ballal SH. Dense Deposit Disease Involving C3 and C4d Deposits. Indian J Nephrol. 2018 Jan-Feb;28(1):61-64. doi: 10.4103/ijn.IJN_164_16. PMID: 29515303; PMCID: PMC5830811.
- Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C, Ghiggeri GM, Ravazzolo R, Savino M, Del Vecchio M, d'Apolito M, Iolascon A, Zelante LL, Savoia A, Balduini CL, Noris P, Magrini U, Belletti S, Heath KE, Babcock M, Glucksman MJ, Aliprandis E, Bizzaro N, Desnick RJ, Martignetti JA. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet. 2000 Sep;26(1):103-5. doi: 10.1038/79063. PMID: 10973259.
- Han KH, Lee H, Kang HG, Moon KC, Lee JH, Park YS, Ha IS, Ahn HS, Choi Y, Cheong HI. Renal manifestations of patients with MYH9-related disorders. Pediatr Nephrol. 2011 Apr;26(4):549-55. doi: 10.1007/s00467-010-1735-3. Epub 2011 Jan 6. PMID: 21210153.
- Ghiggeri GM, Caridi G, Magrini U, Sessa A, Savoia A, Seri M, Pecci A, Romagnoli R, Gangarossa S, Noris P, Sartore S, Necchi V, Ravazzolo R, Balduini CL. Genetics, clinical and pathological features of glomerulonephritis associated with mutations of nonmuscle myosin IIA (Fechtner syndrome). Am J Kidney Dis. 2003 Jan;41(1):95-104. doi: 10.1053/ajkd.2003.50028. PMID: 12500226.
- Gopalakrishnan I, Iskandar SS, Daeihagh P, Divers J, Langefeld CD, Bowden DW, Hicks PJ, Rocco MV, Freedman BI. Coincident idiopathic focal segmental glomerulosclerosis collapsing variant and diabetic nephropathy in an African American homozygous for MYH9 risk variants. Hum Pathol. 2011 Feb;42(2):291-4. doi: 10.1016/j.humpath.2010.07.016. Epub 2010 Nov 13. PMID: 21074826; PMCID: PMC3022108.